John-Michael Gamble
As pharmacotherapy experts, pharmacists are continually updating their knowledge about drug effects. In addition to being knowledge users of research findings, pharmacists increasingly play a larger role in observational studies of drug effects. Observational studies are inherently nonexperimental and, unlike randomized clinical trials (RCTs), do not involve any manipulation (such as randomization) of the treatment and control groups by the investigator.
This article reviews for the practising pharmacist the fundamental design elements and foundational methodologic knowledge for conducting cohort and case–control studies, 2 common and robust observational study designs for elucidating drug–outcome associations. Readers interested in learning about other observational study designs, such as cross-sectional studies, ecological studies, case series, case reports, within-person studies, and quasi-experimental designs, or the critical appraisal of such designs, are referred elsewhere.1–6
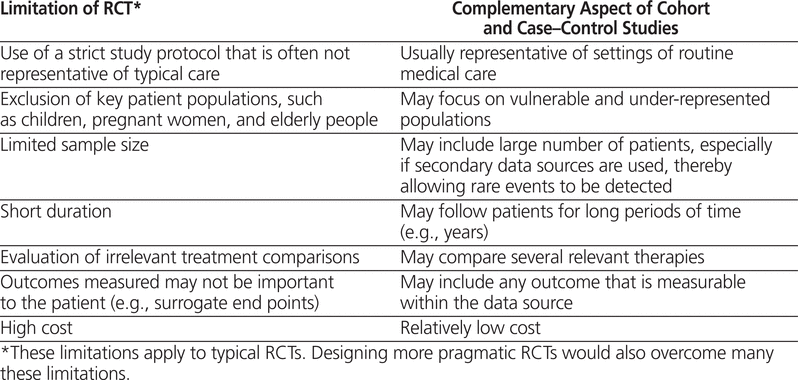
We need well-designed and rigorous cohort and case– control studies because their findings provide knowledge complementary to that garnered from RCTs (Table 1). The design properties of RCTs maximize their ability to estimate the potential causal effects of drugs under ideal circumstances and thereby to estimate the efficacy of those drugs. However, many RCTs involve a relatively limited number of highly selected patients and a limited duration. Indeed, RCTs typically follow patients for only a small fraction of the time that the drug would be used in clinical practice, especially when the medications are for chronic diseases. Moreover, RCTs typically exclude complex patients, they often use irrelevant comparators (e.g., placebo), and they frequently measure outcomes that are not patient-centred (i.e., surrogate end points).7 Although many of these limitations may be overcome by designing more pragmatic RCTs that do indeed measure effectiveness,8 cohort and case–control studies are 2 feasible study design alternatives that address the limitations of RCTs (Table 1) without the considerable financial and human resource costs of pragmatic RCTs.
Table 1.
Limitations of Randomized Clinical Trials (RCTs) Potentially Addressed by Cohort and Case–Control Studies
A cohort is a group of people who share a common experience or characteristic. The term “cohort” first appeared in the medical literature in the 1930s in an article by epidemiologist W H Frost.9 Interestingly, the word “cohort” has military roots, originating from the Latin word “cohors”.10 The term was first used in the Roman military, where a group of 300 to 600 soldiers constituted a cohort.11
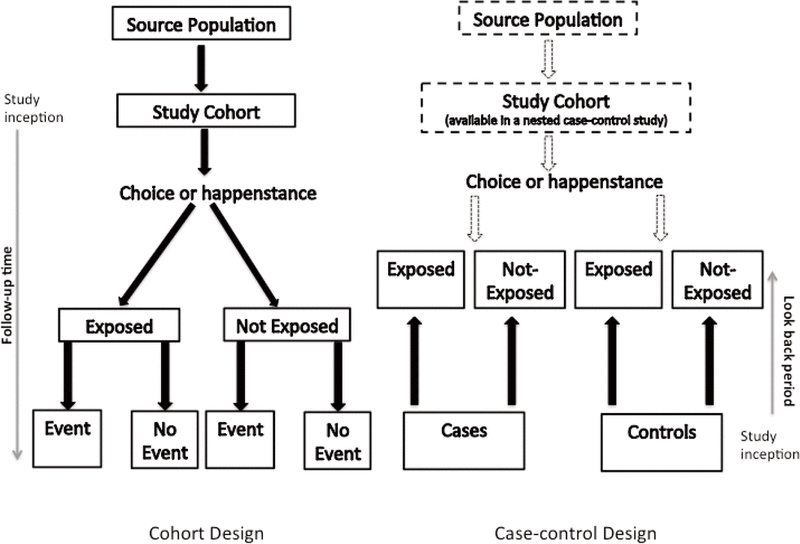
A cohort study compares the experience of 2 or more groups of patients who are followed concurrently forward in time (Figure 1). This prospective tracking, from exposure to outcome, is in fact one of the defining features of a cohort study.11 The temporal sequence involved in following a group of patients who are exposed to a certain factor (the treatment group) and a group of patients who are not exposed to that factor (the control group) is akin to that of a clinical trial, where instead of chance determining a patient’s exposure status (as occurs in an RCT), choice or happenstance determines exposure status.
|
|
||
|
Figure 1. Schematic for the design of cohort and case–control studies. |
||
For any cohort study, a source population must be defined, from which the eligible study cohort is derived through application of various inclusion and exclusion criteria. At a minimum, patients entering the study cohort must be free of the outcome of interest. For example, in a cohort study designed to measure the association between atypical antipsychotics and diabetes mellitus, patients with diabetes would have to be excluded from the study cohort because they are not at risk of the outcome. Often, other restrictions are put in place to minimize the risk of bias. For example, restriction to new users of a medication will ensure avoidance of multiple biases.12 Inclusion of prevalent or current drug users can lead to significant bias because patients who experience early intolerance or adverse effects of a drug may discontinue the drug, and the remaining cohort will consist of a healthier and usually more adherent group.13 Risk that varies over time, whereby new users have a higher risk of an adverse event, has been observed for numerous associations, including those between nonsteroidal anti-inflammatory drugs and upper gastrointestinal bleeding,14 oral contraceptives and venous thromboembolism,15 benzodiazepines and falls,16 and angiotensin-converting enzyme inhibitors and angioedema.17
Once the study cohort has been created, 2 or more exposure groups must be clearly defined, 1 of which must serve as the control or reference group. The reference group should be clinically relevant. For example, in a comparative safety or effectiveness study, patients taking a drug within the same therapeutic class or receiving usual care may serve as the reference group. If clinically and scientifically relevant, a group with no therapeutic exposure may be the reference group. Drug exposure may be measured in terms of persons or person-time (the time for which a person is exposed to a particular drug). Drug exposure is often categorized in a binary fashion (i.e., yes or no), based on either a minimum number of prescription records (e.g., at least 3 records) or a specified duration of exposure (e.g., at least 90 days’ exposure), or a combination of these 2 factors (i.e., cumulative exposure). Irrespective of how exposure is defined, it is essential that follow-up time be properly categorized following entry into the cohort to avoid time-related bias.18 Furthermore, the definition of exposure should be coherent with the study hypothesis. For example, a certain amount of time or a certain dose of drug may be required to elicit an effect, or a drug may continue to have an effect once discontinued (e.g., bisphosphonates). Moreover, decisions about when to discontinue drug exposure must be made. There are 2 common approaches: “as treated”, whereby drug exposure is recorded as being stopped when a person no longer meets the definition of exposure; and “intention-to-treat”, whereby a person is considered exposed from the time of first meeting the study’s exposure definition until experiencing the outcome of interest or the end of the study, irrespective of changes in actual exposure status. There is no consensus on how to best define drug exposures, and hence the definitions of exposure often vary considerably among cohort studies assessing identical drug–outcome associations.
Complete and accurate measurement of the outcome of interest is essential to ensure the validity of study results. When subjective outcome data (e.g., diagnosis of pneumonia) are being collected during the study period, exposure status should be blinded for the outcome assessors and adjudicators, to prevent responder bias. When previously collected data (i.e., secondary data) are being used, investigators should ideally use outcome definitions that have been validated in previous studies. For example, Hux and others19 validated definitions of diabetes by comparing International Classification of Diseases codes obtained from administrative health care databases in Ontario with diagnostic data from primary care charts.
For cohort studies, the drug–outcome association is usually expressed as a relative risk, a relative rate, or a hazard ratio. Advanced statistical techniques are used to account for factors other than the drug exposure of interest that might distort the drug–outcome association. These factors or potential confounders are often handled simultaneously with multivariable regression models. Because these statistical models account for measured variables, it is crucial that the data source capture as many potential confounding variables (or proxies of confounders) as possible. Potential confounders should usually be measured before entry into the cohort, to avoid adjustment for factors in the causal pathway.
One of the major strengths of a cohort study is that the temporal sequence—drug exposure preceding outcome—is explicit in the study design. The incidence of a particular outcome among persons exposed to a certain drug can be directly calculated using a cohort design. Cohort studies are also relatively efficient for studying rare exposures, and multiple outcomes may be assessed for a single exposure. However, cohort studies with long observation periods may be more susceptible to losses to follow-up and to inaccurate measurement of exposures and outcomes. Large numbers of patients may be required to precisely estimate meaningful drug–outcome associations, especially for rare outcomes or outcomes that take a long time to occur.
The first case–control study using the design with which we are familiar today was published in 1926. However, the concept of case–control studies has its origins in the investigation of disease etiologies through detailed histories and examination of patients.20
In a case–control study, a number of cases and noncases (controls) are identified, and the occurrence of one or more prior exposures is compared between groups to evaluate drug–outcome associations (Figure 1). A case–control study runs in reverse relative to a cohort study.21 As such, study inception occurs when a patient experiences an outcome and is thus designated a “case”. A modern conceptual view holds that the case–control study can be thought of as an efficient cohort design. Essentially, patients who would have experienced the outcome of interest in a cohort study are the cases in a case–control study. Similarly, patients who were at risk but did not experience the outcome of interest in a cohort study are the controls in a case–control study. The potential data sources for a case–control study are identical with those for a cohort study, and the investigator may collect data after study inception or may use previously collected data. An extension of the case–control study is the nested case–control study, which is a case–control study conducted within a cohort. Details regarding this design are beyond the scope of this article and are reviewed elsewhere.22,23
The first step in a case–control study is to identify the cases through application of explicitly defined inclusion and exclusion criteria. Ideally, cases should be directly sampled from the source population in a manner that is unrelated to the drug exposures of interest; however, the source population that gave rise to the cases is often unknown and difficult to identify (except in a nested-case control study, where the source population is known). The case-selection process and the data sources from which cases were selected should be described in detail, especially if cases are from a variety of sources, such as hospital and community-based sources. Selecting only hospital-based cases may lead to systematic error related to hospital admission practices, whereby exposed cases may be more likely to be admitted and therefore selected into the study (a phenomenon known as Berksonian bias). Furthermore, only new (incident) cases should be selected, as nonincident cases usually over-represent long-term survivors, and diagnostic practices may change over time, introducing potential bias. When cases are selected from a secondary data source, the case definitions should be supported by previous validation studies.
The selection of controls in a case–control study is fraught with difficulty and is often the source of significant bias. Essentially, the controls should be selected from the same source population as the cases.24 In other words, controls should be at risk of becoming cases and should come from a population with the same exposure distribution as the cases. Multiple controls are usually selected for each case, to increase the statistical efficiency of the study; however, the gains are minimal beyond 3 or 4 controls per case. Nonetheless, modern case–control studies involving large databases often use much higher control–case ratios to maximize study precision. To control for potential confounding, cases and controls are often matched on one or more patient characteristics, such as age or sex (although it may not always be appropriate to match on these variables). The study investigator must be careful not to match on too many factors or on factors that are not confounders, as doing so might lead to overmatching and bias. Furthermore, matching should not involve variables that the investigator is interested in examining in association with an outcome. The selection of controls is one of the most difficult aspects of epidemiologic research, and readers are encouraged to consult additional resources.24–28
Similar to the situation for a cohort study, the drug exposures of interest and their definitions should be clearly specified in the methods. Because exposure in a case–control study is determined after the cases have been identified, a period before occurrence of the case, called the “look-back period” or “look-back window”, must be defined. A comparable look-back period must be defined for the control group. Look-back periods should consider the study hypothesis and thus may vary considerably from one study to another. For example, Abdelmoneim and others29 specified a 120-day look-back period before the date of their cases (patients with acute coronary syndrome) to assess recent exposure to glyburide and gliclazide. Azoulay and others30 specified an exposure window of any time prior to a year before the date of cases in their study evaluating the association between pioglitazone and bladder cancer. If the investigators are collecting exposure data themselves, then outcome status should be blinded to study personnel.
In a case–control study, the odds ratio is the usual measure of association reported. This measure is the ratio of the odds of an exposure between cases and controls and in most cases approximates the relative risk. As in a cohort study, the analytic plan for a case–control study typically involves advanced statistical methods to adjust for multiple potential confounders.
The major strengths of the case–control design are statistical efficiency (i.e., uses fewer data to quantify a drug–outcome association than would be required in a cohort study), efficiency for studying rare outcomes, efficiency for studying conditions with long latency periods, efficiency for handling the time-varying nature of drug exposures, and relatively low cost. The weaknesses of case–control studies include inefficiency for studying rare exposures, difficulty of selecting unbiased controls, and inability to directly calculate incidence rates of outcomes.
Observational studies are methodologically difficult, susceptible to bias and confounding, and difficult to interpret, given the many types of bias potentially at play. For these reasons, observational studies are limited to studying drug–outcome associations and cannot be used to measure the causal effects of drugs. Recent methodologic advances in design and analytic techniques in pharmacoepidemiology have helped to combat the various types of selection bias, information bias, and confounding at play in cohort and case–control studies (see Appendix 1, available online at www.cjhp-online.ca/index.php/cjhp/issue/view/104/showToc). Many of these techniques can account for multiple potential confounders simultaneously. A comprehensive review of these techniques is beyond the scope of this article, but such reviews may be found elsewhere,25,31–33 Bias and confounding result in spurious drug–outcome associations and are introduced at both the design and analysis stages. Appendix 2 (available online at www.cjhp-online.ca/index.php/cjhp/issue/view/104/showToc) illustrates the origin of bias in relation to the cohort design, and Appendix 3 (available online at www.cjhp-online.ca/index.php/cjhp/issue/view/104/showToc) lists common types of bias that occur in cohort and case–control studies of drug effects.
Cohort and case–control studies are powerful approaches for estimating the association between drugs and unintended outcomes34; however, their use for studying the intended effects of drugs has spurred debate in the past and remains controversial today.35–37 This controversy has arisen because the propensity for bias and confounding is much higher when estimating the intended effects of drugs (i.e., benefits).37 This higher propensity for bias is in turn due to the nonrandom nature of prescribing practices and is often referred to as “confounding by the reason for the prescription” or simply “confounding by indication”. Confounding by indication is expected with these types of studies, as it is good medical practice to prescribe intentionally and rationally, as opposed to prescribing according to a random process.38 Some authors strongly recommend against using observational studies to study intended effects, suggesting instead that we consider restricting our research questions to those of unintended effects because confounding by indication introduces uncontrollable bias.31,34,39,40 The literature contains numerous examples of confounding by indication. A most striking example is the distorted 27-fold increased risk of thrombotic events associated with use of warfarin, when in fact warfarin prevents thrombotic events.39 Another example of confounding by indication is the observed relationship between short-acting ß-receptor agonists (e.g., salbutamol) and increased risk of death from asthma.41 Of course confounding by indication is not verifiable, but it must be considered when studying the intended effects of drugs.
Cohort and case–control studies aim to quantitatively estimate the association between a drug exposure and outcome. Before embarking on a cohort or case–control study, the investigators must develop a well-articulated and focused research question.42 Furthermore, the study protocol, including a detailed methodologic and analytic plan, should be consistent with international guidelines.43,44 The study team should have appropriate clinical and methodologic expertise. Clinical expertise is essential for developing exposure and outcome definitions, as well as for understanding the overall clinical context of how the research question fits into the current body of knowledge. Methodologic expertise is critical for ensuring that robust methods are used, to minimize bias and confounding.
To estimate a drug–outcome association in a cohort or case– control study, accurate and comprehensive data must be collected on the drug exposures and outcomes of interest. Study investigators may collect data after study inception or may use previously collected data. The major advantage of prospectively collecting the data (primary data collection) is that the investigators have control over what information is collected; in contrast, when previously collected data are used (secondary data collection), the investigators are limited to the information already collected. Data may often be missing from or inaccurately recorded in secondary data sources, which creates challenges when the data are used for research purposes. Although previously collected data are considered retrospective to study inception, the data themselves are often collected prospectively; therefore, use of the terms “retrospective” and “prospective” may be misleading and usually does not provide any clarity in terms of important design characteristics.25 There are 3 main sources of existing data: administrative data, medical records, and surveys. Special considerations and the advantages and disadvantages of these secondary data sources are discussed elsewhere.45,46 For studying drug effects, secondary data sources are more commonly used than primary data collection, primarily because of gains in time, cost, and statistical efficiency. Furthermore, use of secondary data sources avoids the Hawthorne effect, whereby knowledge of participation in a study changes the behaviour of study participants and may lead to bias.
Pharmacists use knowledge from cohort and case–control studies to inform patients, clinicians, and the general public about drug effects. At a basic level, cohort and case–control studies quantitatively estimate the relation between exposures and outcomes. They represent rigorous study designs for answering drug safety and effectiveness questions, with case–control studies being more prone to bias. The methodologic rigour of cohort and case–control studies evaluating drug–outcome associations is advancing, and approaches are being developed and refined that limit the generation of misleading study results. Indeed, both RCTs and observational studies are necessary, and neither is sufficient to learn about the totality of drug effects in the population.
1. Hartung DM, Touchette D. Overview of clinical research design. Am J Health Syst Pharm. 2009;66(4):398–408.
2. Etminan M, Samii A. Pharmacoepidemiology I: a review of pharmacoepidemiologic study designs. Pharmacotherapy. 2004;24(8):964–9.
3. Schafermeyer K, Hurd D. Research methodology: designing study. J Manag Care Pharm. 1998;4(5):504–14.
4. DiPietro N. Methods in epidemiology: observational study designs. Pharmacotherapy. 2010;30(10):973–84.
5. Shields KM, DiPietro NA, Kier KL. Principles of drug literature evaluation for observational study designs. Pharmacotherapy. 2011;31(2):115–27.
6. Guyatt G, Rennie D, Meade MO, Cook DJ. Users’ guide to the medical literature: a manual for evidence-based clinical practice. 2nd ed. Toronto (ON): McGraw-Hill; 2008.
7. Avorn J. In defense of pharmacoepidemiology—embracing the yin and yang of drug research. N Engl J Med. 2007;357(22):2219–21.
8. Singal AG, Higgins PDR, Waljee AK. A primer on effectiveness and efficacy trials. Clin Transl Gastroenterol. 2014;5(1):e45. 
9. Doll R. Cohort studies: history of the method. I. Prospective cohort studies. Soz Präventivmed. 2001;46(2):75–86.
10. Song JW, Chung KC. Observational studies: cohort and case–control studies. Plast Reconstr Surg. 2010;126(6):2234–42.
11. Grimes DA, Schulz KF. Cohort studies: marching towards outcomes. Lancet. 2002;359(9303):341–5.
12. Ray WA. Evaluating medication effects outside of clinical trials: new-user designs. Am J Epidemiol. 2003;158(9):915–20.
13. Shrank WH, Patrick AR, Brookhart MA. Healthy user and related biases in observational studies of preventive interventions: a primer for physicians. J Gen Intern Med. 2011;26(5):546–50.
14. Yola M, Lucien A. Evidence of the depletion of susceptibles effect in non-experimental pharmacoepidemiologic research. J Clin Epidemiol. 1994; 47(7):731–7.
15. Suissa S, Blais L, Spitzer WO, Cusson J, Lewis M, Heinemann L. First-time use of newer oral contraceptives and the risk of venous thromboembolism. Contraception. 1997;56(3):141–6.
16. Ray WA, Thapa PB, Gideon P. Benzodiazepines and the risk of falls in nursing home residents. J Am Geriatr Soc. 2000;48(6):682–5.
17. Burkhart DG, Brown NJ, Griffin MR, Ray WA, Hammerstrom T, Weiss S. Angiotensin converting enzyme inhibitor-associated angioedema: higher risk in blacks than whites. Pharmacoepidemiol Drug Saf. 1996;5(3):149–54.
18. Suissa S. Immortal time bias in observational studies of drug effects. Pharmacoepidemiol Drug Saf. 2007;16(3):241–9.
19. Hux JE, Ivis F, Flintoft V, Bica A. Diabetes in Ontario: determination of prevalence and incidence using a validated administrative data algorithm. Diabetes Care. 2002;25(3):512–6.
20. Paneth N, Susser E, Susser M. Origins and early development of the case–control study: part 1, early evolution. Soz Präventivmed. 2002; 47(5):282–8.
21. Schulz KF, Grimes DA. Case–control studies: research in reverse. Lancet. 2002;359(9304):431–4.
22. Ernster VL. Nested case–control studies. Prev Med. 1994;23(5):587–90.
23. Etminan M. Pharmacoepidemiology II: the nested case–control study—a novel approach in pharmacoepidemiologic research. Pharmacotherapy. 2004;24(9):1105–9.
24. Wacholder S, McLaughlin JK, Silverman DT, Mandel JS. Selection of controls in case–control studies. I. Principles. Am J Epidemiol. 1992;135(9): 1019–28.
25. Rothman KJ, Greenland S, Lash TL. Modern epidemiology. 3rd ed. New York (NY): Lippincott Williams & Wilkins; 2008.
26. Grimes DA, Schulz KF. Compared to what? Finding controls for case– control studies. Lancet. 2005;365(9468):1429–33.
27. Wacholder S, Silverman DT, McLaughlin JK, Mandel JS. Selection of controls in case–control studies. II. Types of controls. Am J Epidemiol. 1992; 135(9):1029–41.
28. Wacholder S, Silverman DT, McLaughlin JK, Mandel JS. Selection of controls in case–control studies. III. Design options. Am J Epidemiol. 1992; 135(9):1042–50.
29. Abdelmoneim AS, Eurich DT, Gamble JM, Johnson JA, Seubert JM, Qiu W, et al. Risk of acute coronary events associated with glyburide compared with gliclazide use in patients with type 2 diabetes: a nested case–control study. Diabetes Obes Metab. 2014;16(1):22–9.
30. Azoulay L, Yin H, Filion KB, Assayag J, Majdan A, Pollak MN, et al. The use of pioglitazone and the risk of bladder cancer in people with type 2 diabetes: nested case–control study. BMJ. 2012;344:e3645.
31. McMahon AD. Approaches to combat with confounding by indication in observational studies of intended drug effects. Pharmacoepidemiol Drug Saf. 2003;12(7):551–8.
32. Klungel OH, Martens EP, Psaty BM, Grobbee DE, Sullivan SD, Stricker BH, et al. Methods to assess intended effects of drug treatment in observational studies are reviewed. J Clin Epidemiol. 2004;57(12):1223–31.
33. Schneeweiss S, Suissa S. Chapter 47: Advanced approaches to controlling confounding in pharmacoepidemiologic studies. In: Strom B, Kimmel S, Henness S, editors. Pharmacoepidemiology. 5th ed. Chichester (UK): Wiley-Blackwell; 2012. p. 868–92.
34. McMahon AD, MacDonald TM. Design issues for drug epidemiology. Br J Clin Pharmacol. 2000;50(5):419–25.
35. Byar DP. Why data bases should not replace randomized clinical trials. Biometrics. 1980;36(2):337–42.
36. Pocock SJ, Elbourne DR. Randomized trials or observational tribulations? N Engl J Med. 2000;342(25):1907–9.
37. Vandenbroucke JP, Psaty BM. Benefits and risks of drug treatments: how to combine the best evidence on benefits with the best data about adverse effects. JAMA. 2008;300(20):2417–9.
38. Laupacis A, Mamdani M. Observational studies of treatment effectiveness: some cautions. Ann Intern Med. 2004;140(11):923–4.
39. Miettinen OS. The need for randomization in the study of intended effects. Stat Med. 1983;2(2):267–71.
40. Vandenbroucke JP. When are observational studies as credible as randomised trials? Lancet. 2004;363(9422):1728–31.
41. Ernst P, Spitzer WO, Suissa S, Cockcroft D, Habbick B, Horwitz RI, et al. Risk of fatal and near-fatal asthma in relation to inhaled corticosteroid use. JAMA. 1992;268(24):3462–4.
42. Tully MP. Research: articulating questions, generating hypotheses, and choosing study designs. Can J Hosp Pharm. 2014;67(1):31–4.
43. Guide on methodological standards in pharmacoepidemiology. Revision 2. EMA/95098/2010. London (UK): European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP); 2013 Jun 18 [cited 2014 Aug 1]. Available from: www.encepp.eu/standards_and_guidances/documents/ENCePPGuideMethStandardsPE_Rev2.pdf
44. International Society for Pharmacoepidemiology. Guidelines for good pharmacoepidemiology practices (GPP). Pharmacoepidemiol Drug Saf. 2007;18(2):200–8.
45. Hennessy S. Use of health care databases in pharmacoepidemiology. Basic Clin Pharmacol Toxicol. 2006;98(3):311–3.
46. Harpe S. Using secondary data sources for pharmacoepidemiology and outcomes research. Pharmacotherapy. 2009;29(2):138–53
John-Michael Gamble is supported by a New Investigator Award in drug safety and effectiveness from the Canadian Institutes of Health Research and a Clinician Scientist Award from the Canadian Diabetes Association.
Competing interests: None declared.
Canadian Journal of Hospital Pharmacy , VOLUME 67 , NUMBER 5 , September-October 2014