Joane Y Tang, Tony K L Kiang, Mary H H EnsomABSTRACT
Background
Coadministration of lorazepam and valproic acid is identified by tertiary references as causing a major drug interaction that requires therapy modification and dosage adjustments. The proposed mechanism involves inhibition of lorazepam glucuronidation via direct inhibition of uridine 5′-diphosphate-glucuronosyltransferase enzymes by valproic acid. However, the clinical significance of this interaction is unclear.
Objectives
To identify site-specific practices and assess clinical responses to the interaction between valproic acid and lorazepam.
Methods
A chart review was conducted for patients over 18 years of age who were admitted, from September 2008 to September 2014 inclusive, to the psychiatry or neurology service at Vancouver General Hospital, Vancouver, British Columbia, and who received concomitant valproic acid and lorazepam therapy.
Results
Of the 30 patients included in the chart review, 12 (40%) received an intervention. A total of 8 (27%) patients experienced an adverse drug reaction (ADR), such as drowsiness and dizziness. Seven of these 8 patients were among those who received an intervention. The mean dosage (± standard deviation) of lorazepam was 4.2 ± 1.2 mg per day among patients who experienced an ADR and less than 2 mg per day among those who did not experience an ADR.
Conclusions
The current recommendation from tertiary drug references is to reduce the dose of lorazepam by 50% when this drug is coadministered with valproic acid. However, this recommendation could not be validated through an analysis of patients exposed to this interaction in the clinical setting or through a review of the literature. Further clinical and pharmacokinetic studies are required to determine whether concurrent treatment with lorazepam and valproic acid should be considered as causing a major drug interaction. Until more data are available, clinicians should remain cognizant of the potential for a drug–drug interaction and should use the lowest effective dose of lorazepam when this drug is administered concomitantly with valproic acid.
KEYWORDS: valproic acid, lorazepam, pharmacokinetics, drug-drug interaction, uridine 5′-diphosphate-glucuronosyltransferases
RÉSUMÉ
Contexte
Selon des sources tertiaires, la prise concomitante de lorazépam et d’acide valproïque causerait une interaction médicamenteuse grave nécessitant un changement au traitement et des ajustements posologiques. Le mécanisme qui sous-tend cette interaction est l’inhibition de la glucuronoconjugaison du lorazépam au moyen de l’inhibition directe des enzymes uridine 5′-diphosphate glucuronosyltransférases par l’acide valproïque. Cependant, la signification clinique de cette interaction est inconnue.
Objectifs
Déterminer quelles sont les pratiques particulières à l’établissement hospitalier et évaluer les réponses cliniques à l’interaction entre l’acide valproïque et le lorazépam.
Méthodes
On a mené une analyse des dossiers médicaux des patients de plus de 18 ans qui, entre septembre 2008 et septembre 2014 inclusivement, ont été admis au service de psychiatrie ou de neurologie de l’Hôpital général de Vancouver, à Vancouver en Colombie-Britannique, et ont reçu en même temps l’acide valproïque et le lorazépam.
Résultats
Parmi les 30 patients dont le dossier médical a été retenu pour l’analyse, 12 (40 %) ont eu droit à une intervention. Un total de 8 (27 %) patients ont subi une réaction indésirable aux médicaments (RIM), comme de la somnolence et des étourdissements. Sept de ces 8 patients appartenaient au groupe qui a eu droit à une intervention. La dose quotidienne moyenne de lorazépam était de 4,2 ± 1,2 mg chez les patients qui ont subi une RIM et de moins de 2 mg chez ceux qui n’en ont pas subi.
Conclusions
Des sources tertiaires sur les médicaments recommandent actuellement de réduire la dose de lorazépam de 50 % lorsque ce médicament est administré conjointement avec l’acide valproïque. Or, cette recommandation n’a pu être validée à l’aide d’une revue de la littérature ou d’une analyse des dossiers des patients ayant été exposés à une telle interaction médicamenteuse en milieu clinique. De plus amples études cliniques et pharmacocinétiques sont nécessaires pour déterminer si la prise concomitante de lorazépam et d’acide valproïque peut être considérée comme une cause d’interaction médicamenteuse grave. D’ici à ce qu’ils disposent de plus de données probantes, les cliniciens doivent demeurer conscients du potentiel d’interaction médicament-médicament et utiliser la plus faible dose efficace de lorazépam lorsque ce médicament est pris en concomitance avec l’acide valproïque.
MOTS CLÉS: acide valproïque, lorazépam, pharmacocinétique, interaction médicament-médicament, uridine 5′-diphosphate glucuronosyltransférases
Lorazepam is a short-acting benzodiazepine1 often used concurrently with valproic acid, a broad-spectrum antiepileptic drug, in the treatment of epilepsy and psychiatric conditions such as bipolar disorder.2 Valproic acid is available in an acid form and an enteric-coated salt form (divalproex sodium); the latter provides delayed release and absorption from the intestine, but the extent of absorption from the 2 formulations is equivalent.3,4 Lorazepam and valproic acid are both metabolized via glucuronidation by uridine 5′-diphosphate-glucuronosyltransferase (UGT) enzymes.1,5 Valproic acid is both a known inhibitor and a substrate of UGT, with glucuronidation accounting for about 33% to 60% of its metabolism.6–9 Lorazepam is also extensively metabolized by the same UGT enzymes that are inhibited by valproic acid.10,11
The proposed pharmacokinetic (PK) interaction between lorazepam and valproic acid is believed to be due to the inhibition of glucuronidation, presumably by direct inhibition of UGT enzymes by valproic acid.12 In vitro studies in rat and human liver tissue have shown that valproic acid can decrease glucuronidation activity, resulting in an increase in the area under the plasma concentration–time curve (AUC) by 15% to 18%.10,11 However, the reported values of the inhibitor constant for the inhibition of lorazepam glucuronidation by valproic acid are much greater than concentrations that are usually achieved in vivo.11,13 This situation alludes to the limitations of in vitro UGT inhibition studies, given that the inhibitory effects may not occur in humans.7
Theoretically, inhibition of lorazepam glucuronidation may prolong its sedative effects and pose potential safety concerns. Several case reports of stupor and coma have suggested the drug combination as the potential cause.14,15 However, it is unclear whether pharmacodynamic effects and clinical outcomes are correlated with a drug–drug interaction at the PK level.
This study was a chart review conducted at an urban teaching hospital (Vancouver General Hospital, Vancouver, British Columbia) with the purpose of assessing clinical responses to the drug interaction between valproic acid and lorazepam in relation to site-specific practices when these 2 drugs were administered concurrently. The primary objective was to determine the frequency and types of intervention when valproic acid and lorazepam were prescribed concomitantly. Secondary outcomes included the rationale for therapy intervention and the adverse outcomes that patients experienced.
A chart review was conducted with a convenience sample of 30 patients over 18 years of age. To be included, patients had to have been admitted to the psychiatry or neurology service of Vancouver General Hospital, Vancouver, British Columbia, from September 2008 to September 2014, with concomitant prescription of valproic acid and lorazepam. Patients with a prescription for lorazepam to be taken as needed but who did not receive any lorazepam (as per the medication administration record) were excluded. Patients with end-stage renal disease (estimated glomerular filtration rate < 15 mL/min or on dialysis) were also excluded. The following information was obtained from the patients’ charts: demographic characteristics; baseline characteristics; total daily doses of valproic acid and lorazepam; most responsible psychiatric diagnosis; concurrent medications taken in hospital; number of, types of, and rationale for interventions performed when valproic acid and lorazepam were prescribed concomitantly; and any adverse outcomes experienced.
Means, standard deviations, and ranges were calculated for continuous data (using Microsoft Excel 2011, version 14.1.3, Microsoft Corporation, Redmond, Washington).
Ethics approval was obtained from the University of British Columbia Research Ethics Board.
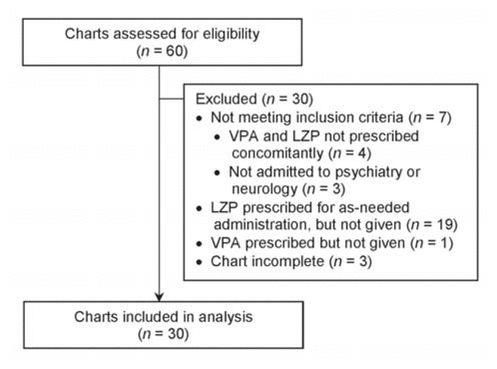
Of 60 patients whose charts were screened, 30 met the inclusion criteria (Table 1). The most common reason for exclusion was prescription of lorazepam to be taken as needed with no doses actually administered, as per the medication administration record (Figure 1). Eight (27%) of the patients were female, and the mean age was 41.4 years (range 19–76) (Table 2). Of the 30 patients reviewed, 18 (60%) did not receive any intervention. The remaining 12 patients (40%) received an intervention, the most common being dosage adjustments, discontinuation of lorazepam, and restarting of valproic acid at a lower dosage (Figure 2). Three patients underwent more than one type of intervention.
Table 1 Inclusion and Exclusion Criteria
|
|
||
|
Figure 1 Flow diagram of chart review to identify study participants. LZP = lorazepam, VPA = valproic acid. |
||
Table 2 Baseline Characteristics of Patients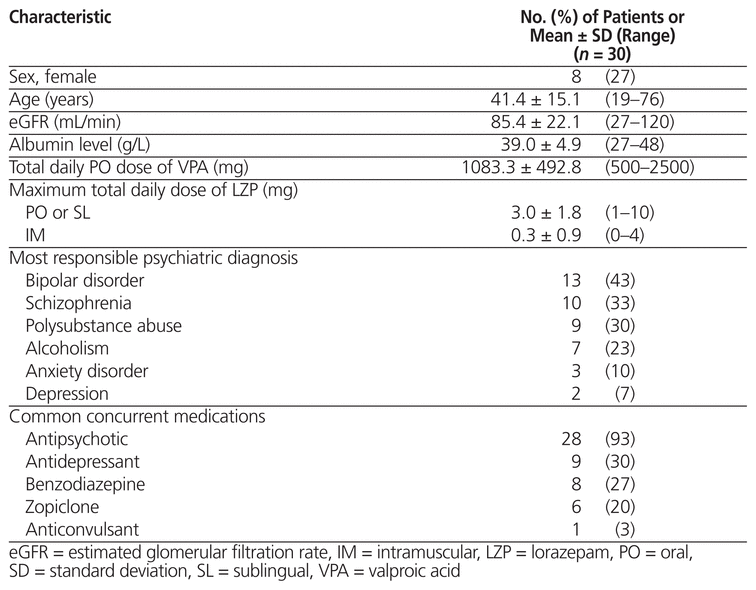
|
|
||
|
Figure 2 Types of intervention in 30 study patients who were receiving concomitant lorazepam (LZP) and valproic acid (VPA) therapy. |
||
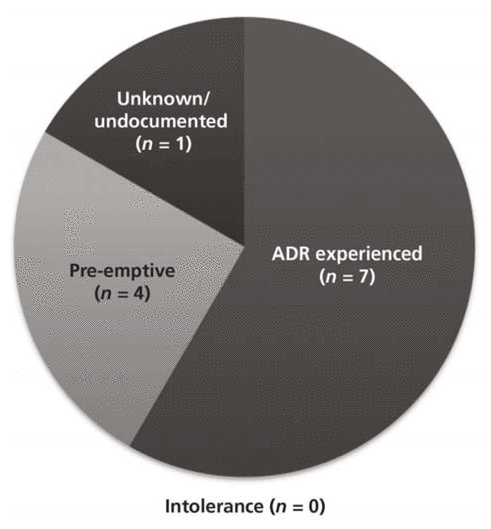
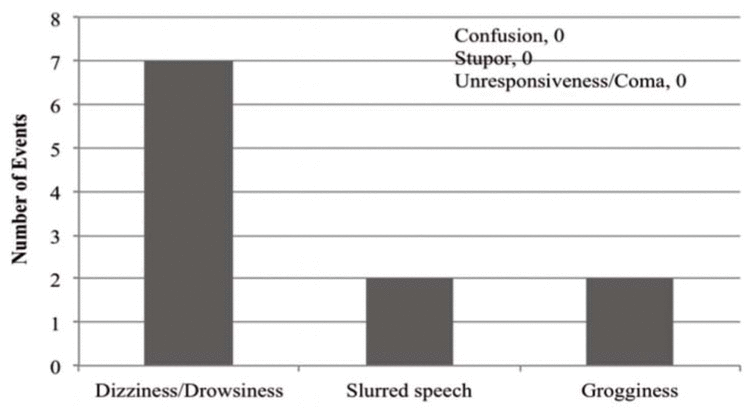
The most frequent rationale for therapy intervention was occurrence of an adverse drug reaction (ADR) (Figure 3). A total of 8 patients (27%) were reported to have experienced one or more ADRs (Figure 4), of whom 7 received an intervention; one patient did not receive any intervention despite the ADR. Five patients who did not experience any ADR also received an intervention. Two patients experienced more than one ADR. Among patients who experienced ADRs, the mean dosage of oral or intramuscular lorazepam was 4.2 ± 1.2 mg per day. Conversely, most of the patients who did not experience ADRs (16 of 22) received less than 2 mg per day.
|
|
||
|
Figure 3 Rationale for interventions in the 30 study patients. ADR = adverse drug reaction. |
||
|
|
||
|
Figure 4 Adverse outcomes experienced by 8 patients receiving concomitant lorazepam and valproic acid therapy. Two of the patients experienced more than one adverse outcome. |
||
All 30 of the patients received concurrent antipsychotic medications, and 15 (50%) received medications from more than one class. The most commonly used classes of medication were benzodiazepines and antidepressants. Five (63%) of the 8 patients who experienced an ADR received a single first-generation antipsychotic (i.e., loxapine or methotrimeprazine), and 2 patients received 2 or more first-generation antipsychotics (Table 3). One patient received an intramuscular dose of zuclopenthixol decanoate, which has a duration of action of about 2 weeks and a half-life (t1/2) of 19 days.16
Table 3 Concurrent Medications Taken by Patients Who Experienced Adverse Drug Reactions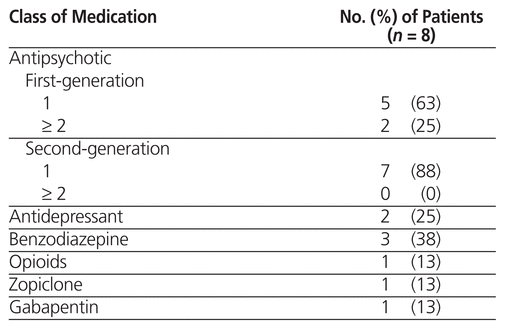
To the authors’ knowledge, this chart review appears to be the first study to report the clinical effects of the potential drug–drug interaction between lorazepam and valproic acid. The identification and management of drug interactions are essential components of patient care.
This chart review showed that a small number of patients (8 of 30) experienced a transient, non–life-threatening ADR, most commonly dizziness or drowsiness. The mean dosage of lorazepam received by patients who experienced ADRs was about 4.2 mg per day, compared with less than 2 mg per day for patients with no reported ADRs. It is difficult to differentiate whether the observed ADRs were a result of the compounded sedative effect of the higher lorazepam dose alone or the prolongation of effects of lorazepam secondary to PK inhibition by valproic acid. From the data in this retrospective study, we are unable to draw any concrete conclusions regarding the likelihood that the drug interaction was solely responsible for the observed ADRs. However, it can be expected that the higher the dose of lorazepam, the greater the amount of the drug that will accumulate in the presence of valproic acid inhibition, assuming that clearance remains constant.
A thorough literature search was conducted to review the PK evidence (i.e., absorption, distribution, metabolism, and excretion) surrounding the drug interaction between lorazepam and valproic acid, and to further assess whether the recommendations from tertiary resources are valid. Four PK studies that compared the combination of valproic acid and lorazepam with lorazepam alone were identified.9,17–19 The data from those studies are summarized in Table 4.
Table 4 Summary of Pharmacokinetic Studies Involving Coadministration of Valproic Acid (VPA) and Lorazepam (LZP)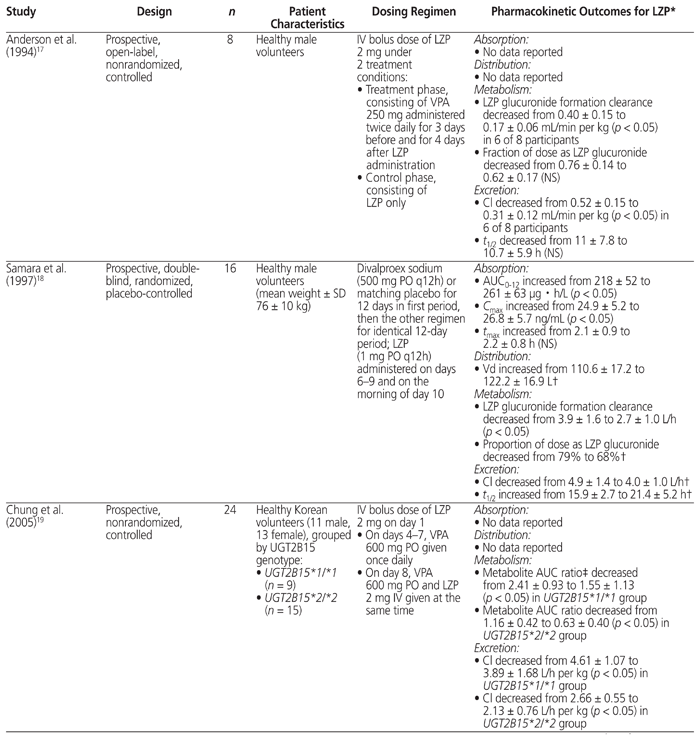

According to available evidence, neither the absorption of lorazepam nor its volume of distribution appear to be affected by coadministration with valproic acid.18 All 4 studies showed that valproic acid cotreatment inhibited the metabolism of lorazepam, as indicated by an observed decrease in either lorazepam glucuronide formation clearance17,18 or the metabolite AUC ratio.9,19 With regard to elimination, a decrease in plasma clearance of lorazepam was observed in all 4 studies.9,17–19
Some limitations in the interpretation of data from clinical drug interaction studies deserve mention. For example, in addition to UGT induction or inhibition, formation clearance may also be affected by clearance or AUC of the parent drug and other drug-metabolizing enzymes or transport proteins.7
Of the few relevant publications identified in the literature search, only one described a randomized study.18 That study used multiple doses of lorazepam,18 whereas most studies used only a single dose,9,17,19 which may not truly reflect clinical practice. None of the studies objectively assessed the clinical outcome of the drug interaction. Moreover, there was significant heterogeneity in study design (e.g., dosage, route of administration, treatment duration, period of coadministration with valproic acid, patient population, and PK outcomes reported).
The current chart review was limited by its retrospective design and the small sample of patients. Data were collected by reviewing medication records, which may be affected by incomplete, missing, or inaccurate documentation. Furthermore, clinical situations could be interpreted incorrectly if charting was vague. In addition, chart reviews do not fully capture the complete clinical decision-making process. The high incidence of polypharmacy, particularly with other medications having sedative properties, complicates interpretation of the data. Information on objective measures such as the Glasgow Coma Scale was not collected because although some patients had a baseline score, scores were not always documented at subsequent stages of therapy, making it difficult to assess any changes that might have occurred. In addition, any changes in the score could have resulted from existing medical conditions as well as from medications. Another limitation of this chart review was the lack of information about laboratory parameters (e.g., valproic acid concentration, ammonia concentration, liver function tests, complete blood count), data that may assist in delineating whether the observed neuropsychiatric ADRs were due to valproic acid alone or the drug–drug interaction. Although objective data such as laboratory values may be helpful, such values are not expected to change dramatically in the presence of concomitant lorazepam and valproic acid therapy, because the proposed mechanism of the interaction would likely cause changes in the concentrations of lorazepam and lorazepam glucuronide, rather than the concentration of valproic acid.
Patients with end-stage renal disease and reduced renal function were excluded from this chart review because these conditions may lead to an accumulation of lorazepam glucuronide and may indirectly affect the concentration of the parent compound.20 The rising concentration of the glucuronide would theoretically inhibit further metabolite formation from the parent drug, thereby prolonging the plasma t1/2 of the latter and increasing its concentration.21
One way to better evaluate the likelihood of the drug interaction would be to implement dechallenge and rechallenge of the precipitating drug.22 This approach may not always be feasible in the clinical setting. It would also be difficult to evaluate the process and results of dechallenge and rechallenge in a retrospective study if details were not documented clearly at the time of care. In this particular chart review, we were unable to identify any cases involving dechallenge of valproic acid with improvement in clinical symptoms of ADRs. To fully assess the valproic acid–lorazepam interaction, more reliable PK data are required from well-designed randomized, controlled PK studies, involving a larger population and multiple doses of medications to mimic clinical practice.
Tertiary drug references23 identify the concurrent administration of lorazepam with valproic acid as a “major” drug interaction supported by “excellent reliability”, and recommend a 50% dose reduction, as per the product monograph of lorazepam.20 However, there is a lack of clinical and literature support for this recommendation. Although a small number of patients in the current study experienced transient and minor ADRs, a causal relationship with the drug interaction could not be determined. Although the literature search provided some evidence to support a metabolism-mediated PK drug interaction, it was difficult to draw conclusions, given the small number of studies to date, their methodological flaws, the limited data on PK parameters, and the lack of objective clinical outcomes. Hence, more studies (preferably prospective, randomized, and controlled) are needed to determine whether valproic acid increases the sedative effects of lorazepam or prolongs the effects of lorazepam through reduced clearance.
Until future studies can provide more supportive evidence to demonstrate that the interaction between lorazepam and valproic acid is a major concern, clinicians should remain cognizant of the potential drug interaction and use the lowest effective dose of lorazepam when it is administered concomitantly with valproic acid. There should also be close patient monitoring to avoid oversedation and serious central nervous system depression.
1 Riss J, Cloyd J, Gates J, Collins S. Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol Scand. 2008;118(2):69–86.
2 Swann AC. Valproic acid: clinical efficacy and use in psychatric disorders. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic drugs. 5th ed. Philadelphia (PA): Lippincott Williams & Wilkins; 2002. p. 828–36.
3 Valproate sodium, valproic acid, divalproex sodium. In: AHFS: drug information [database on Internet]. Bethesda (MD): American Society of Health-System Pharmacists; 2014 [cited 2014 Jul 19]. Accessed through: MedicinesComplete at https://www-medicinescomplete-com.ezproxy.library.ubc.ca/mc/index.htm [university-based subscription access].
4 Gugler R, von Unruh GE. Clinical pharmacokinetics of valproic acid. Clin Pharmacokinet. 1980;5(1):67–83.
5 Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16(10):695–714.
6 de Leon J. Glucuronidation enzymes, genes and psychiatry. Int J Neuropsychopharmacol. 2003;6(1):57–72.
7 Kiang TKL, Ensom MHH, Chang TKH. UDP-glucuronosyltransferases and clinical drug–drug interactions. Pharmacol Ther. 2005;106(1):97–132.
8 Ethell BT, Anderson GD, Burchell B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem Pharmacol. 2003;65(9):1441–9.
9 Chung JY, Cho JY, Yu KS, Kim JR, Lim KS, Sohn DR, et al. Pharmacokinetic and pharmacodynamic interaction of lorazepam and valproic acid in relation to UGT2B7 genetic polymorphism in healthy subjects. Clin Pharmacol Ther. 2007;83(4):595–600.
10 Stingl JC, Bartels H, Viviani R, Lehmann ML, Brockmöller J. Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: a quantitative systematic review. Pharmacol Ther. 2014;141(1):92–116.
11 Uchaipichat V, Suthisisang C, Miners JO. The glucuronidation of R- and S-lorazepam: human liver microsomal kinetics, UDP-glucuronosyltransferase enzyme selectivity, and inhibition by drugs. Drug Metab Dispos. 2013; 41(6):1273–84.
12 Lin JH, Wong BK. Complexities of glucuronidation affecting in vitro–in vivo extrapolation. Curr Drug Metab. 2002;3(6):623–46.
13 Dutta S, Zhang Y, Selness DS, Lee LL, Williams LA, Sommerville KW. Comparison of the bioavailablity of unequal doses of divalproex sodium extended-release formulation relative to the delayed-release formulation in healthy volunteers. Epilepsy Res. 2002;49(1):1–10.
14 Uz Y, Hariri A, Ünübol H, Bilici M. Stupor due to possible interaction between lorazepam and valproic acid: report of two cases. Turkish J Psychiatry. 2012;23(4):281–3.
15 Lee SA, Lee JK, Heo K. Coma probably induced by lorazepam–valproate interaction. Seizure. 2002;11(2):124–5.
16 Zuclopenthixol. In: Lexi-Drugs Online [database on Internet]. Hudson (OH): Wolters Kluwer; [cited 2016 Apr 3]. Available from: http://online.lexi.com/action/home [subscription required to access content].
17 Anderson GD, Gidal BE, Kantor ED, Wilensky AJ. Lorazepam–valproate interaction: studies in normal subjects and isolated perfused rat liver. Epilepsia. 1994;35(1):221–5.
18 Samara EE, Granneman RG, Witt GF, Cavanaugh JH. Effect of valproate on the pharmacokinetics and pharmacodynamics of lorazepam. J Clin Pharmacol. 1997;37(5):442–50.
19 Chung JY, Cho JY, Yu KS, Kim JR, Jung HR, Lim KS, et al. Effect of the UGT2B15 genotype on the pharmacokinetics, pharmacodynamics, and drug interactions of intravenous lorazepam in healthy volunteers. Clin Pharmacol Ther. 2005; 77(6):486–94.
20 Ativan (lorazepam) [product monograph]. Montréal (QC): Pfizer Canada, Inc; 2014 [cited 2015 Jun 15]. Available from: www.pfizer.ca/sites/g/files/g10017036/f/201505/PM_Ativan_Clean_E_Level_3_10-Oct-2014.pdf
21 Verbeeck R, Tjandramaga TB, Verberckmoes R, de Schepper PJ. Biotransformation and excretion of lorazepam in patients with chronic renal failure. Br J Clin Pharmacol. 1976;3(6):1033–9. 
22 Horn JR, Hansten PD, Chan LN. Proposal for a new tool to evaluate drug interaction cases. Ann Pharmacother. 2007;41(4):674–80.
23 Lorazepam and valproic acid. In: Lexicomp Interact [database on Internet]. Hudson (OH): Wolters Kluwer; [cited 2015 Feb 19]. Available from: https://www-myrxtx-ca.ezproxy.library.ubc.ca/NEW/TOOL/DI [university-based subscription access].
Competing interests: None declared. ( Return to Text )
Funding: None received. ( Return to Text )
Canadian Journal of Hospital Pharmacy, VOLUME 70, NUMBER 3, May-June 2017