Apixaban Concentration with and without Coadministration of Carbamazepine: A Case with No Apparent Interaction
Norelle Evanger, Artur Szkotak, Linda Stang, Tammy J Bungard
INTRODUCTION
The direct-acting oral anticoagulants (DOACs) have many advantages over warfarin. In particular, apixaban has rapid onset (1–3 h) and offset (half-life 8–15 h) of action, with predictable dosing, which precludes the need for routine coagulation monitoring.1 In contrast to warfarin, which has numerous drug–drug interactions, apixaban has fewer reported drug–drug interactions.2,3 Apixaban is primarily metabolized by the cytochrome P450 isozymes CYP3A4 and CYP3A5 and is a substrate for the P-glycoprotein (P-gp) transport protein.4 Drugs that are strong inhibitors or inducers of both CYP3A4 and P-gp are reported to increase and decrease apixaban concentrations, respectively. The manufacturer’s product monograph states that strong inhibitors of both these pathways are contraindicated in combination with apixaban, whereas those that are strong inducers (such as carbamazepine) should “generally be avoided” given the likelihood of reduced apixaban concentrations4; however, there are no specific data describing this interaction.5
Carbamazepine is a strong inducer of CYP3A4 and P-gp, and the extent to which it lowers apixaban concentrations is not known.2,3 Although testing for apixaban concentration is not widely available, most large centres have the ability to monitor heparin (anti-Xa) levels. Moreover, a linear relation has been reported between measured values on a heparin activity assay and apixaban concentrations in plasma.6–8 We report here a case in which apixaban concentrations calculated from locally measured heparin levels were compared with calibrated apixaban concentrations measured by a laboratory in Hamilton, Ontario, both during concomitant carbamazepine–apixaban therapy and after discontinuation of carbamazepine. We also reflect on the available literature that informed our case management.
CASE REPORT
A 64-year-old, 103-kg man was referred to our anticoagulation clinic for management of newly started warfarin therapy following diagnosis of atrial flutter.* The CHADS2 score was 5 (based on a history of heart failure with ejection fraction of 30%, hypertension, type 2 diabetes mellitus, and transient ischemic attack). The patient also had diverticulosis, chronic kidney disease (serum creatinine 154 µmol/L, creatinine clearance 49 mL/min), a traumatic brain injury, vertebral basilar artery syndrome, depression, and epilepsy (for which carbamazepine 200 mg bid had been prescribed). DOAC therapy had been avoided because of the concomitant use of carbamazepine.
The patient’s other medications were furosemide 40 mg bid, metoprolol 50 mg bid, spironolactone 25 mg daily, acetylsalicylic acid 81 mg daily, atorvastatin 80 mg daily, valsartan 320 mg daily, repaglinide 0.5 mg tid, and bupropion XL 300 mg daily. All of his medications, except warfarin, were blister packed for 2-week periods, and the prescriptions were consistently filled by the pharmacy. The primary contact to ensure adherence with warfarin therapy was the patient’s daughter.
In a recent 1-month period, the patient had been admitted to hospital twice for exacerbation of heart failure. Despite gradual escalation of the warfarin dose beyond what had been required before the hospital admissions to achieve therapeutic international normalized ratio (INR), with weekly INR assessment after the second admission by the anticoagulation clinic, the patient’s INR remained subtherapeutic for 5 weeks. The institution’s heart function clinic discontinued the warfarin therapy and prescribed apixaban 5 mg bid instead. Given the recent inadequacy of warfarin control and the manufacturer’s recommendation to “generally avoid” (as opposed to contraindicating) this drug combination, because of uncertainty about the effect on apixaban concentration, the anticoagulation clinic pursued an assessment of apixaban trough and peak concentrations. After 10 weeks of concomitant carbamazepine–apixaban administration, the family doctor discontinued the carbamazepine and initiated valproic acid 250 mg tid, as a way to avoid the purported carbamazepine–apixaban interaction. Twelve weeks later, measurement of apixaban concentration was repeated.
DISCUSSION
This case features 3 aspects of care delivery: assessment of apixaban concentration using a locally developed strategy (given that apixaban assays are not widely available), review of available literature addressing apixaban concentration in patients with atrial fibrillation, and factors considered in developing the rationale for management in this case.
Local Measurement of Heparin and Calculation of Apixaban Concentrations
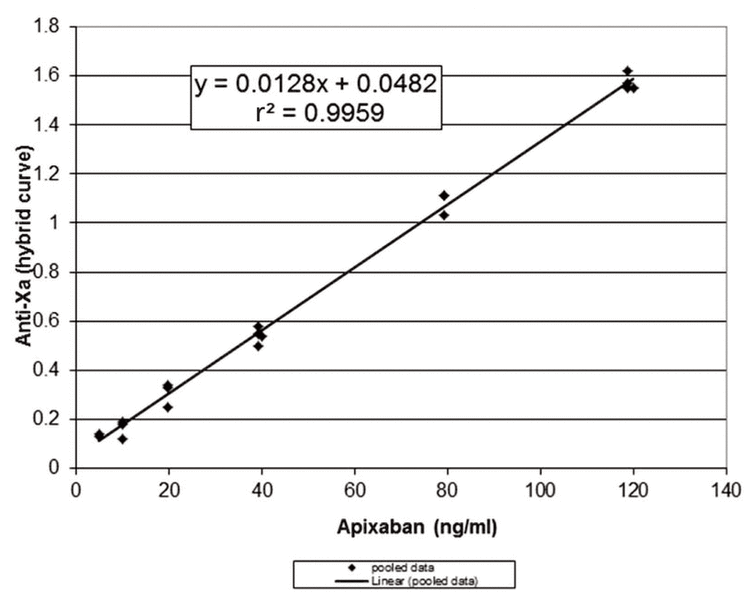
Given the strong linear correlation between anti-factor Xa and apixaban concentration, an anti-Xa assay is appropriate for measuring apixaban concentrations.6–8 However, apixaban-calibrated anti-Xa assays are not widely available. Therefore, the University of Alberta Hospital laboratory performed in vitro spiking experiments using known quantities of apixaban (Bristol-Myers-Squibb, Plainsboro, New Jersey) added to normal pooled plasma, with anti-Xa levels measured using the Biophen heparin 6 anti-Xa based assay (Aniara, Mason, Ohio) calibrated to measure unfractionated heparin and low-molecular-weight heparin in units per millilitre, using a hybrid curve. These assays were replicated over 3 days and showed good precision (standard deviation 0.04 U/mL) with excellent linear correlation (r2 = 0.9959) (Figure 1). Despite the strong linear correlation shown by these in vitro experiments, we were uncertain how apixaban concentration and anti-Xa would correlate in vivo.
| |
|
|
|
Figure 1 Relation between anti-Xa and apixaban, based on in vitro spiking experiments using known quantities of apixaban added to normal pooled plasma and anti-Xa measured by the Biophen heparin 6 assay (calibrated to measure unfractionated heparin and low-molecular-weight heparin). The curve shows pooled data over 3 days.
|
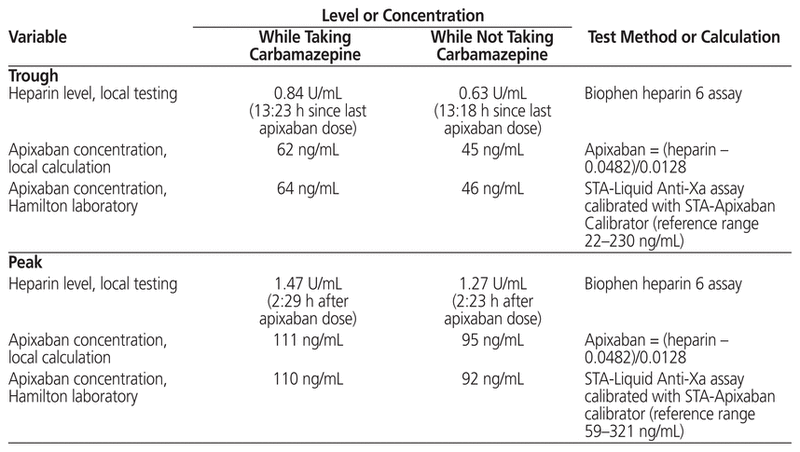
Samples for measurement of peak and trough concentrations were drawn following 10 days of concomitant apixaban–carbamazepine administration and again 12 weeks after discontinuation of carbamazepine (Table 1). Anti-Xa levels were measured locally, and a calculated prediction of apixaban concentration was obtained using the formula derived from the in vitro spiking data, as described above. These results were compared with those obtained from a reference laboratory in Hamilton, Ontario, which measures apixaban concentration with the STA-Liquid Anti-Xa Assay calibrated with STA-Apixaban Calibrator (Diagnostica Stago, Asnières Sur Seine, France). The apixaban concentrations calculated at the University of Alberta Hospital and those reported from the Hamilton laboratory were essentially the same (Table 1). Therefore, we concluded that the local Biophen heparin 6 anti-Xa based assay, with calculation of apixaban concentrations using the formula created with the in vitro data, was reliable for this case.
Table 1 Anti-Xa Levels and Apixaban Concentrations for a Patient Who Also Received Carbamazepine
Patient’s Apixaban Concentrations and Literature Review
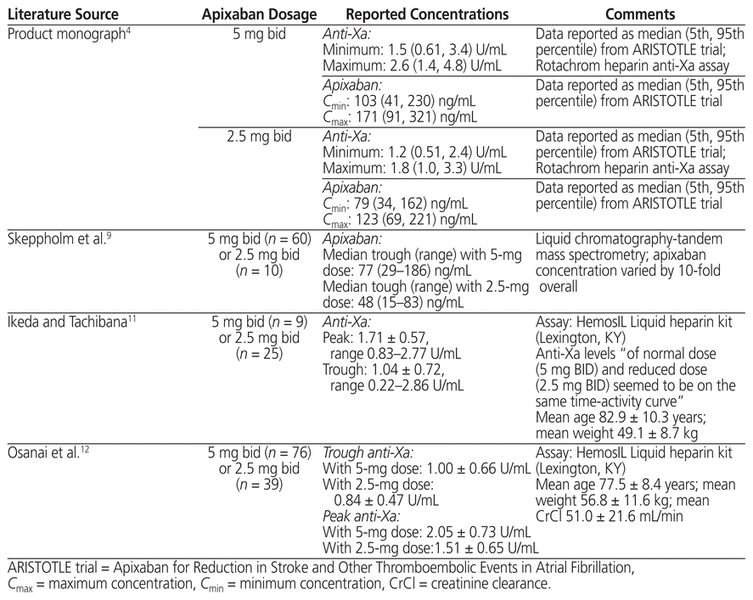
The patient’s apixaban trough concentrations (64 and 46 ng/mL while taking and not taking carbamazepine, respectively) and peak concentrations (110 and 92 ng/mL, respectively) fell within the reported reference ranges, albeit toward the lower end reported for apixaban 5 mg bid therapy and closer to the median concentrations reported for an apixaban 2.5 mg bid regimen4 (Tables 1 and 2). A coagulation centre in Stockholm conducted a study of trough apixaban concentrations using liquid chromatography-tandem mass spectrometry to identify the typical exposure of apixaban in patients with atrial fibrillation.9 Sixty patients receiving apixaban 5 mg bid had a median apixaban trough concentration of 77 ng/mL (range 29–186 ng/mL), whereas 10 patients receiving apixaban 2.5 mg bid had a median trough concentration of 48 ng/mL (range 15–83 ng/mL). Our patient’s apixaban concentration reasonably aligned with that of patients receiving apixaban 5 mg bid. In another study of consecutive patients with atrial fibrillation for whom apixaban was prescribed at 4 anticoagulation clinics, apixaban concentrations were higher than what was achieved in the current case, with the mean peak concentrations reported by 3 of the clinics being 242, 227, and 133 ng/mL.10 Mean trough concentrations were similarly higher, at 110, 127, and 91 ng/mL. The authors reported wide ranges for each mean concentration reported, owing to inter-individual variability. Two studies of Japanese patients with atrial fibrillation11,12 reported higher heparin levels both at peak (1.71 ± 0.57 U/mL and 2.05 ± 0.73 U/mL) and trough (1.04 ± 0.72 U/mL and 1.00 ± 0.66 U/mL) than those observed in this case (1.47 U/mL and 0.84 U/mL, respectively). These investigators used a different commercial assay (HemosIL liquid heparin kit, Instrumentation Laboratory, Lexington, Kentucky) and did not report calculated apixaban concentrations. The higher heparin levels may be attributable, in part, to the use of a different commercial assay, as well as to patient factors: the Japanese patients had lower body weight (mean 49 and 58 kg) and older age (mean 83 and 73 years) than the patient described here (weight 103 kg and age 64 years).
Table 2 Literature Review of Apixaban Measurements for Patients with Atrial Fibrillation
Case Management
The patient’s apixaban trough (46 ng/mL) and peak (92 ng/mL) concentrations 12 weeks after discontinuation of carbamazepine were lower than those reported while taking carbamazepine (64 and 110 ng/mL, respectively) which was unexpected, given the offset of enzyme and P-gp induction. When the second set of apixaban concentrations were calculated, factors that were altered included an increase in serum creatinine to 186 µmol/L (creatinine clearance 40 mL/min) and initiation of valproic acid. Given the apparent decline in creatinine clearance, we would have anticipated, if anything, an increase in apixaban concentration. In addition, there has been no mention of concern with the concomitant use of valproic acid and apixaban.4,13 In vitro studies yielded conflicting evidence, with one reporting inhibition of CYP3A4 by valproic acid14 and the other reporting induction.15 In terms of an effect on P-gp, in vitro data from rat liver and human tumour cells demonstrated that valproic acid induced P-gp expression,16 which implies a potential for reducing apixaban concentration. Overall, however, the likelihood of a meaningful effect of valproic acid on apixaban concentration is low, given that strong induction of both P-gp and CYP3A4 is necessary to warrant concern.4 Of note, in the current case, we did not restart carbamazepine following reassessment of apixaban concentrations (which would have been akin to the practice of restarting a drug, to reproduce signs/symptoms, in cases of adverse events) because the patient remained seizure-free while taking valproic acid.
Ultimately, we elected not to up-titrate the patient’s apixaban dose, despite apixaban concentrations being at the lower end of ranges stated in the product monograph and reported in the literature for most patients with atrial fibrillation. Our rationale for this decision was based on several factors. First, clinical outcomes have not been correlated with apixaban concentrations, and only average concentration ranges have been reported, with no established therapeutic range.8 Previous authors who suggested measurement of DOAC concentrations in unique situations have offered no insight or guidance regarding the interpretation of these concentrations or ways to adjust the DOAC therapy, because no data exist. Second, although the inter-individual variability with apixaban concentrations is less than that for rivaroxaban and dabigatran, data indicate there is still a 7- to 10-fold variation in plasma concentrations for patients with nonvalvular atrial fibrillation.9,10,17 Although our patient’s concentrations were on the low end, they still fell within reported ranges. Third, this patient had recent exacerbations of heart failure (which made warfarin control challenging), and creatinine clearance calculated by the Cockcroft–Gault equation was 40–49 mL/min. Had further deterioration ensued, having apixaban concentrations at the lower end of the range offered more leeway to avoid exceeding the upper limit of reported ranges. Finally, the patient was taking a lower-than-usual dose of carbamazepine (400 mg daily; typical dosing range 1000–4000 mg daily), which implied a lower level of induction.18 One study of 29 patients with epilepsy supports the notion of a dose-dependent relation of enzyme induction with carbamazepine through demonstration of an increase in clearance of antipyrine and urinary excretion of d-glucaric acid.19 These data are limited, however, in that the carbamazepine doses were higher (mean 14.0 ± 6.0 mg/kg per day) than the dose in this case, and P-gp induction was not assessed.
CONCLUSION
We have reported a case in which apixaban concentrations were assessed both during and after discontinuation of carbamazepine therapy, and local heparin hybrid calibrated anti-Xa assays were used to calculate apixaban concentrations, with these calculated concentrations being consistent with those reported by an external laboratory. Apixaban concentrations after discontinuation of carbamazepine were lower than expected, most likely indicating a lack of drug interaction in this particular patient, who was taking a relatively low dose of carbamazepine. Although the patient’s trough and peak concentrations were on the lower end of reported ranges, we elected to not change the apixaban dosing. This case report suggests that empirically adjusting DOAC doses based on potential interactions may be dangerous. More information specific to target DOAC concentrations and patient outcomes is needed to guide clinical decisions.
References
1 Dempfle CE. Direct oral anticoagulants—pharmacology, drug interactions, and side effects. Semin Hematol. 2014;51(2):89–97.

2 Hellwig T, Gulseth M. Pharmacokinetic and pharmacodynamics drug interactions with new oral anticoagulants: what do they mean for patients with atrial fibrillation? Ann Pharmacother. 2013;47(11):1478–87.
3 Nutescu E, Chuatrisorn I, Hellenbart E. Drug and dietary interactions of warfarin and novel oral anticoagulants: an update. J Thromb Thrombolysis. 2011;31(3):326–43.
4 Eliquis™ product monograph. Kirkland (QC): Pfizer Canada; 2016 Jun 16.
5 Stöllberger C, Finsterer J. Interactions between non-vitamin K oral anticoagulants and antiepileptic drugs. Epilepsy Res. 2016;126:98–101.
6 Dale BJ, Chan NC, Eikelboom JW. Laboratory measurement of the direct oral anticoagulants. Br J Haematol. 2016;172(3):315–36.
7 Frost C, Song Y, Barrett YC, Wang J, Pursley J, Boyd RA, et al. A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin Pharmacol Adv Appl. 2014;6:179–87.
8 Martin K, Moll S. Direct oral anticoagulant drug level testing in clinical practice: a single institution experience. Thromb Res. 2016;143:40–4.

9 Skeppholm M, Al-Aieshy F, Berndtsson M, Al-Khalili F, Rönquist-Nii Y, Söderblom L, et al. Clinical evaluation of laboratory methods to monitor apixaban treatment in patients with atrial fibrillation. Thromb Res. 2015; 136(1):148–53.
10 Testa S, Tripodi A, Legnani C, Pengo V, Abbate R, Dellanoce C, et al. Plasma levels of direct oral anticoagulants in real life patients with atrial fibrillation: results observed in four anticoagulation clinics. Thromb Res. 2016;137:178–83.
11 Ikeda K, Tachibana H. Clinical implication of monitoring rivaroxaban and apixaban by using anti-factor Xa assay in patients with non-valvular atrial fibrillation. J Arrhythmia. 2016;32(1):42–50.
12 Osanai H, Ajioka M, Masutomi T, Kuwayama T, Ishihama S, Sakamato Y, et al. Measurement of anti-factor Xa activity in pateints on apixaban for non-valvular atrial fibrillation. Circ J. 2015;79(12):2584–90.
13 Depakene® valproic acid [product monograph]. Etobicoke (ON): BGP Pharma ULC; 2014 Dec 22 [revised 2016 Jan 8; cited 2017 Jun 7]. Available from: www.mylan.ca/-/media/mylanca/documents/english/product%20pdf/pdfs%20dec%202015/depakene-pm-2016.01.08.pdf
14 Wen X, Wang JS, Kivistö KT, Neuvonen PJ, Backman JT. In vitro evaluation of valproic acid as an inhibitor of human cytochrome P450 isoforms: preferential inhibition of cytochrome P450 2C9 (CYP2C9). Br J Clin Pharmacol. 2001;52(5):547–53.
15 Cerveny L, Svecova L, Anzenbacherova E, Vrzal R, Staud F, Dvorak Z, et al. Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos. 2007;35(7):1032–41.
16 Eyal S, Lamb JG, Smith-Yockman M, Yagen B, Fibach E, Altschuler Y, et al. The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat livers. Br J Pharmacol. 2006; 149(3):250–60.
17 Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013;29(7 Suppl):S24–S33.
18 Dilantin™ product monograph. Kirkland (QC): Pfizer Canada; 2016 Feb 22.
19 Perucca E, Hedges A, Makki KA, Ruprah M, Wilson JF, Richens A. A comparative study of the relative enzyme inducing properties of anticonvulsant drugs in epileptic patients. Br J Clin Pharmacol. 1984;18(3):401–10.
Norelle Evanger, BScPharm, is with Pharmacy Services, University of Alberta Hospital, Alberta Health Services, Edmonton, Alberta.
Artur Szkotak, MD, PhD, is with Laboratory Services, University of Alberta Hospital, Alberta Health Services, Edmonton, Alberta.
Linda Stang, MLT, is with Laboratory Services, University of Alberta Hospital, Alberta Health Services, Edmonton, Alberta
Tammy J Bungard, BSP, PharmD, is with the Division of Cardiology, University of Alberta, Edmonton, Alberta.
Competing Interests: Tammy Bungard has received honoraria from Bayer and Bristol Myers Squibb-Pfizer and has also received unrestricted grants from Bristol Myers Squibb-Pfizer and Bayer, all for activities unrelated to this article. Artur Szkotak received a grant from Alexion Pharmaceuticals for work unrelated to this article. Norelle Evanger and Linda Stang have no competing interests to report. (
Return to Text
)
Address correspondence to: Dr Tammy J Bungard, EPICORE Centre, 362 Heritage Medical Research Centre, University of Alberta, Edmonton AB T6G 2S2, e-mail: tammy.bungard@ualberta.ca
(Return to Top)
*The patient’s family provided verbal consent for publication of this case report. (
Return to Text
)
Funding: None received. (
Return to Text
)
Canadian Journal of Hospital Pharmacy, VOLUME 70, NUMBER 6, November-December 2017