Karen Lingertat-Walsh, Shirley Law, Scott E WalkerABSTRACT
Background
Domperidone liquid for oral administration is not commercially available in Canada, but is needed for patients who cannot swallow intact tablets.
Objective
To evaluate the stability of domperidone 5 mg/mL suspensions prepared in Oral Mix vehicle and stored, for up to 91 days, in amber polyvinylchloride (PVC) bottles, amber glass bottles, or amber polyethylene terephthalate (PET) bottles at 4°C or 25°C or in polypropylene oral syringes at 25°C.
Methods
Three separate 300-mL batches of domperidone suspension 5 mg/mL were prepared with Oral Mix vehicle. Fifty-millilitre aliquots of the suspension were stored in 100-mL bottles (amber PVC, amber glass, or amber PET). Half of the bottles of each type were stored at 25°C and half at 4°C. On study days 0, 1, 2, 4, 7, 10, 14, 21, 28, 35, 42, 49, 63, 77, and 91, domperidone concentration was determined, with a validated reverse-phase, stability-indicating liquid chromatographic method, in samples drawn from each type of container stored at each temperature. In addition, 1.5-mL aliquots of a fourth 100-mL batch of suspension were stored in 3-mL oral syringes at 25°C and were tested on the same study days.
Results
The concentration of domperidone in all study samples remained above 93% of initial concentration after storage for 91 days. The percent remaining on day 91, based on fastest degradation rate (as represented by the lower limit of the 95% confidence interval [CI]), was at least 92.3% for suspensions stored at 4°C in PVC, glass, and PET bottles. With storage at 25°C, suspensions in PVC and glass bottles retained more than 90% of initial concentration, whereas suspensions in PET bottles and plastic syringes retained 88.9% and 88.0% of initial concentration, respectively.
Conclusions
Because suspensions of domperidone in PET bottles and oral syringes retained less than 90% of their initial concentration on day 91 (based on the 95% CI), it is suggested that such suspensions be stored at 4°C or 25°C in any bottle type or syringe with an assigned beyond-use date not exceeding 75 days.
KEYWORDS: domperidone, stability, suspension
RÉSUMÉ
Contexte
La dompéridone sous forme liquide pour administration orale n’est pas disponible sur le marché au Canada, mais elle est nécessaire pour les patients incapables d’avaler des comprimés entiers.
Objectif
Évaluer la stabilité de suspensions de dompéridone de 5 mg/mL préparées dans l’excipient Oral Mix et conservées jusqu’à 91 jours dans des flacons de polychlorure de vinyle (PVC) ambré, de verre ambré ou de polyéthylène téréphtalate (PET) ambré à 4 °C ou à 25 °C ou dans des seringues orales de polypropylène à 25 °C.
Méthodes
Trois différents lots de 300 mL de suspension de dompéridone de 5 mg/mL ont été préparés à l’aide de l’excipient Oral Mix. Des aliquotes de 50 mL de la suspension ont été entreposées dans des flacons de 100 mL de PVC ambré, de verre ambré ou de PET ambré. La moitié des flacons de chaque type était conservée à 25 °C et l’autre moitié était conservée à 4 °C. Aux jours 0, 1, 2, 4, 7, 10, 14, 21, 28, 35, 42, 49, 63, 77 et 91 de l’étude, les concentrations de dompéridone ont été évaluées sur des échantillons tirés de chaque type de flacons conservés aux deux températures à l’aide d’une épreuve validée mesurant la stabilité par chromatographie liquide en phase inverse. De plus, des aliquotes de 1,5 mL provenant d’un quatrième lot de suspension ont été entreposées dans des seringues orales de 3 mL à 25 °C et ont été analysées aux mêmes jours.
Résultats
Les concentrations de dompéridone dans l’ensemble des échantillons de l’étude conservaient plus de 93 % de la concentration initiale après un entreposage de 91 jours. Le pourcentage restant au jour 91, selon le taux de dégradation le plus rapide (correspondant à la limite inférieure de l’intervalle de confiance (IC) de 95 %), atteignait au moins 92,3 % pour les suspensions entreposées à 4 °C dans les flacons de PVC, de verre et de PET. Lorsqu’elles étaient entreposées à 25 °C, les suspensions contenues dans les flacons de PVC ou de verre conservaient plus de 90 % de la concentration initiale alors que les suspensions contenues dans les flacons de PET ou les seringues de plastique conservaient respectivement 88,9 % et 88,0 % de la concentration initiale.
Conclusions
Comme les suspensions entreposées dans les flacons de PET et les seringues orales conservaient moins de 90 % de leur concentration initiale au jour 91 (selon l’IC de 95 %), on suggère d’entreposer les suspensions de dompéridone à 4 °C ou à 25 °C dans n’importe lequel contenant en l’accompagnant d’une date limite d’utilisation ne dépassant pas 75 jours.
MOTS CLÉS: dompéridone, stabilité, suspension
Domperidone suspension for oral administration is not commercially available in Canada, yet this formulation is needed for patients, especially children, who cannot swallow tablets. To date, only one study concerning the stability of domperidone suspensions has been published.1 That study determined that 1 mg/mL and 10 mg/mL suspensions of this drug in OraBlend vehicle were stable for 91 days with storage in polyvinylchloride (PVC) bottles at 25°C and 4°C. However, there are no data for storage of domperidone suspensions in glass containers, polyethylene terephthalate (PET) containers, or oral syringes. Furthermore, our analysis of typical doses (unpublished data) suggested that a strength of 5 mg/mL, for which there are no data, would be desirable.
This study was conducted to extend the data of Ensom and others1 by evaluating the stability of domperidone in amber PVC, amber glass, and amber PET containers and in 3-mL clear polypropylene oral syringes. Another purpose of this study was to evaluate Oral Mix, a dye-free vehicle (osmolality 1231 mOsm/kg) produced by a different manufacturer. These stability data will be useful for pharmacies wishing to compound a domperidone suspension that is easy to prepare, that has a reasonable concentration (allowing administration of convenient volumes), and that is stable for at least 30 days.
The objectives of this study were to first determine the physical suitability of dye-free Oral Mix as a vehicle for domperidone suspensions and then to determine the physical and chemical stability of 5 mg/mL domperidone suspensions in Oral Mix, with storage in amber PVC bottles, amber glass bottles, amber PET bottles, and clear polypropylene oral syringes for up to 91 days.
Before the stability study, a separate physical study was undertaken to determine the suitability of the Oral Mix vehicle for domperidone suspensions, with regard to ease of compounding and absence of undesirable physical characteristics (e.g., unpleasant odour, caking). All samples were examined immediately after preparation for odour, taste, colour, pH (with a model PHi 510 digital pH meter, Beckman Coulter, Fullerton, California), and ease of resuspension. Samples were stored at either 25°C or 4°C and were examined periodically over 91 days for presence of clumping, ease of resuspension, and changes in odour, taste, colour, and pH. The refrigerated samples were permitted to equilibrate to room temperature before measurement of pH. In addition, all samples used in the stability study (described below) were examined immediately after preparation and on each defined study day for clumping, ease of resuspension, odour, taste, and colour.
The liquid chromatographic system consisted of an isocratic solvent delivery pump (model P4000, Thermo Separation Products, San Jose, California), which pumped a solution of 100% potassium phosphate dibasic (Fisher Scientific, Fair Lawn, New Jersey) adjusted to pH 7 with phosphoric acid. On each study day, the strength of the mobile phase was adjusted to achieve a retention time for domperidone of 3.5 min through a 3.9 cm × 300 mm reversed-phase LC-18 column (Nova Pak, Waters Scientific, Toronto, Ontario) at 1.0 mL/min. Samples of 4 μL were introduced into the liquid chromatographic system using an auto-injector (WISP 712, Waters Scientific). The column effluent was monitored with a variable-wavelength ultraviolet–visible spectrum (UV–VIS) detector (UV 6000, Thermo Separation Products, Fremont, California) at 245 nm. The signal from the detector was integrated and recorded with a chromatography data system (ChromQuest, version 5.0, Thermo Fisher Scientific Inc, Nepean, Ontario).
Following development of the chromatographic system for domperidone, the suitability of the method for use as a stability-indicating assay was tested by analyzing samples of domperidone that had been subjected to accelerated degradation. Two 0.5 mg/mL samples of domperidone (purity > 98%; Sigma Aldrich Canada Co, Oakville, Ontario; product D122, lot 059K4711V) were prepared. The first sample, prepared in water, was adjusted to pH 1.95 (with 1N hydrochloric acid; Fisher Scientific), placed in a 10-mL glass vial, and incubated at 95°C for 200 min. A sample was drawn from the vial every 15 min, of which a 4-μL sample was injected directly into the liquid chromatography system. The second sample, prepared in water and methanol, was adjusted to pH 11.5 (with 1N sodium hydroxide; Fisher Scientific), placed in a 10-mL glass vial, and incubated at 95°C for 227 min. A sample was drawn from the vial every 15 min, of which a 4-μL sample was injected directly into the liquid chromatography system.
The UV–VIS detector was capable of evaluating the UV–VIS spectrum of the chromatographic column effluent every 0.2 s, thus allowing evaluation of the UV-VIS purity of an eluting peak. Changes in the UV–VIS spectrum over the elution profile of the peak of interest would indicate that the peak is contaminated, that the chromatographic method does not separate domperidone from its degradation products, and that the method is therefore unsuitable. However, if (1) the UV–VIS profile does not change during the elution profile of the peak of interest, (2) the UV–VIS spectrum during the elution profile of the peak of interest is identical with that of a sample of known purity (> 98%), and (3) the drug of interest can be degraded to a measurable extent, with both conditions 1 and 2 remaining true during the evaluation of condition 3, the chromatographic system can be judged as stability-indicating.
The chromatograms obtained from each of the degraded domperidone samples were inspected for the appearance of additional peaks, and the domperidone peak was compared between samples for changes in concentration, retention time, and peak shape (by means of electronic overlay and numeric calculation of tailing). The UV spectral purity of the domperidone peak in chromatograms of the degraded samples was compared with the spectrum of the authentic, undegraded sample of domperidone obtained at time 0. These procedures met or exceeded published and accepted standards.2–4
A sample of the Oral Mix vehicle, with and without domperidone, was assayed to ensure that the vehicle did not interfere with the chromatographic assay.
Once assurance of the specificity of the analytical method had been completed, the validation phase was performed, during which accuracy and reproducibility of the standard curves were evaluated over a 5-day period, and system suitability criteria (theoretical plates, tailing and retention times) were developed to ensure consistent chromatographic performance on each study day.5 On each validation day, 10 mg of domperidone (Sigma Aldrich Canada Co; product D122, lot 059K4711V) was accurately weighed and dissolved in methanol to prepare a 1 mg/mL stock solution. This stock solution was further diluted with water to make standards of 0.094, 0.188, 0.375, and 0.750 mg/mL. Then 4 μL of each standard, the 1 mg/mL stock solution, and a blank were chromatographed in duplicate, to create the standard curve. The range of the calibration curve encompassed the diluted test concentration of the domperidone samples. In addition, 3 quality control (QC) solutions (0.125, 0.250, and 0.500 mg/mL) were prepared on each validation day and chromatographed in duplicate. The concentrations of the QC samples were also determined from the standard curve and compared with the known concentrations.
Within-day and between-day errors were assessed by the coefficients of variation of the peak areas of both the QC samples and the standards.
Domperidone suspensions (5 mg/mL) were prepared, according to the procedure outlined in Appendix 1, from 10-mg tablets (Ranbaxy Pharmaceuticals Canada Inc, Mississauga, Ontario; lot 659899) in Oral Mix vehicle (Medisca Pharmaceutique Inc, Saint-Laurent, Quebec; lot K0248M; product specifications are available in the manufacturer’s safety data sheet6). To test storage in bottles, 3 separate 300-mL batches were prepared. Each batch was divided into 50-mL aliquots for placement in the following containers: 6 amber 100-mL PVC bottles (Richards Packaging, Mississauga, Ontario), 6 amber 100-mL glass bottles (Beatson Clark, Rotherham, South Yorkshire, England; distributed by Richards Packaging), and 6 amber 100-mL PET bottles (Eastman Chemical Company, Kingsport, Tennessee; distributed by Jones Packaging, Brampton, Ontario). Each bottle was half-filled, which allowed airspace above the suspension. To test storage in syringes, a fourth 100-mL batch was prepared; from this, 1.5-mL aliquots were drawn up into 3-mL clear oral polypropylene syringes (PreciseDose dispenser system, Medisca Inc; lot 48287/A; syringe specifications include details about polydimethylsiloxane lubricant7). Three bottles of each type were placed in a refrigerator at 4°C, with protection from light. The remaining 3 bottles of each type were stored at 25°C with exposure to ambient fluorescent light. All of the syringes were stored at 25°C, protected from light with a brown UV-protective bag (item MP-320-28; Pharmasystems, Markham, Ontario). These conditions were designed simulate the preparation, use, and storage of suspensions likely to be encountered during clinical practice.
Appendix 1 Compounding instructions for domperidone 5 mg/mL*
After initial compounding on day 0, and subsequently on days 1, 2, 4, 7, 10, 14, 21, 28, 35, 42, 49, 63, 77, and 91, the test containers (bottles and syringes) were shaken well; then, 1 mL of suspension was withdrawn by pipette from each bottle (i.e., the 3 bottles of each type stored at each temperature) and from 3 of the oral syringes stored at room temperature. These 1-mL samples were diluted to 10 mL with methanol, and each diluted sample was then mixed well before centrifugation for 10 min at 2000 rpm. Then, 4 μL of the supernatant was injected into the chromatography column. Each sample was analyzed in duplicate on the day of sampling using the validated liquid chromatographic system with UV detection at 245 nm. The area under the domperidone peak was subjected to least-squares linear regression, and the actual domperidone concentration in each sample was determined by interpolation from the standard curve and correction by the dilution factor.
On each study day, a standard curve was prepared using standards with concentrations as described in the section “Assay Validation” (above) and a blank. In addition, 3 QC samples were prepared, at the same concentrations as described in the section “Assay Validation”. Each standard and QC sample was chromatographed in duplicate on each study day.
For the analytical method, within-day and between-day errors were assessed by the coefficients of variation of the peak areas of both the QC samples and the standards (during both the assay validation and study periods). After determination of the coefficient of variation of the assay, a power calculation showed that duplicate injection had the ability to distinguish between concentrations that differed by at least 10% within each individual container.8,9 On each day of the study, means and coefficients of variations were calculated for replicate analyses (i.e., each sample assayed in duplicate) of the 3 samples for each combination of container type and temperature.
Analysis of variance and multiple linear regression were used to test differences in concentration on different study days, in different containers, and at different storage temperatures. The 5% level was used as the a priori cutoff for significance.
The percent of initial concentration remaining was analyzed by linear regression, with a 95% confidence interval (CI) being constructed around the slope of the curve for percent remaining as a function of study day. The lower limit of this CI was deemed to represent the estimate of fastest degradation rate, with 95% confidence, and the intersection of this rate and 90% of the initial concentration was used to determine the recommended beyond-use date (BUD).
Concentrations were considered within acceptable limits if the measured concentration on a given study day was greater than 90% of the initial (day 0) concentration, and the concentration on that study day, estimated from the fastest degradation rate with 95% confidence, exceeded 90% of the initial (day 0) concentration.
During the preliminary physical study of domperidone suspensions in Oral Mix, ease of compounding was noted. The drug was well suspended, the suspension itself was not thick or viscous, and it was easy to pour. Some settling occurred during storage, but redispersion occurred easily with shaking. No caking or clumping occurred in any suspension stored at either 4°C or 25°C, as determined by ease of pouring and absence of residue upon visual inspection of the bottom of bottles after the suspension was poured into a graduated cylinder. All suspensions were white in colour, were of a smooth consistency, had a sweet cherry odour, and tasted bitter. The pH of the suspensions stored at both 4°C and 25°C ranged between 4.33 and 4.48 for the duration of the preliminary physical study. Similar physical properties were observed during the 91-day stability study.
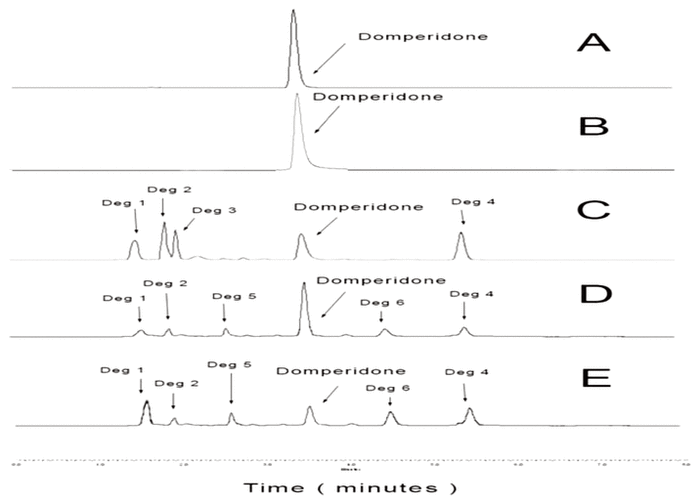
During the accelerated degradation study, consistent degradation of domperidone was observed in both the acidic (pH 1.95) and basic (pH 11.5) samples (Figure 1). Under acidic conditions, 25.32% of the original concentration remained after 200 min, and 4 degradation products were observed (eluting at 1.60, 1.85, 2.00, and 5.50 min). Under basic conditions, 19.84% of the original concentration remained after 227 min, and 5 degradation products were observed (eluting at 1.60, 1.85, 2.60, 4.60, and 5.50 min). Domperidone eluted at 3.50 minutes, and the degradation products did not interfere with domperidone quantification. As a result of the chromatographic separation of the degradation products from domperidone and the similarity of the UV spectrum between an authentic domperidone standard (245 nm) and domperidone in a degraded sample, it was concluded that this analytical method was stability-indicating.
|
|
||
|
Figure 1 Representative chromatograms of domperidone: (A) 0.5 mg/mL in aqueous solution, immediately after preparation; (B) 0.5 mg/mL in aqueous solution, after storage in a polyvinylchloride bottle for 91 days at room temperature (25°C); (C) 0.5 mg/mL in aqueous solution, adjusted to a pH of 1.95 (hydrochloric acid), after incubation at 95°C for 200 min, with 25.32% of the initial concentration remaining; (D) 0.5 mg/mL in water and methanol, adjusted to a pH of 11.5 (sodium hydroxide), after incubation at 95°C for 31 min, with 73.93% of initial concentration remaining; and (E) 0.5 mg/mL in water and methanol, adjusted to a pH of 11.5 (sodium hydroxide), after incubation at 95°C for 227 min, with 19.84% of initial concentration remaining. A total of 6 degradation products are evident in chromatograms C, D, and E, eluting at 1.60 min (designated Deg 1), 1.85 min (Deg 2), 2.00 min (Deg 3), 2.60 min (Deg 5), 4.60 min (Deg 6), and 5.50 min (Deg 4). These degradation products were not evident in samples from the stability study. |
||
Furthermore, the Oral Mix vehicle did not interfere with the domperidone assay. During the 91-day stability study, inspection of chromatograms did not reveal any of the degradation products observed during the accelerated degradation studies (Figure 1, chromatogram B).
Regression analysis of the peak area of domperidone versus the concentration of each domperidone standard demonstrated linearity over the range of concentrations tested, with coefficient of determination (r2) of at least 0.9990 (n = 5).
Analysis of the standard curves and QC samples during the validation study indicated that domperidone concentrations were measured accurately. The standards and QC samples showed less than 3.22% deviation from expected concentration over the validation period. Within-day variation in the slope (the reproducibility, as measured by the coefficient of variation) averaged 0.80% for the standards and 1.51% for the QC samples. Between-day analytical reproducibility, as measured by the coefficient of variation, averaged 1.24% for the standards and 2.18% for the QC samples. Accuracy, based on absolute deviation from the known concentration, averaged 1.84% for the standards and 3.22% for the QC samples.
Regression analysis of the peak area of domperidone versus the concentration of each domperidone standard demonstrated linearity over the range of concentrations tested, with coefficients of determination (r2) of at least 0.9989 (n = 15).
Within-day variation in concentration, as measured by the coefficient of variation, averaged 1.79% for the standards and 2.50% for the QC samples. Between-day analytical reproducibility, as measured by the coefficient of variation, averaged 2.52% for the standards and 5.28% for the QC samples. Accuracy, based on the absolute deviation from the known concentration, averaged 2.46% for the standards and 5.04% for the QC samples. Between-day reproducibility of the study samples, based on the standard deviation of the regression, averaged 2.35%. Therefore, it can be concluded that domperidone was measured accurately and reproducibly on each day of the study, which indicates that differences of 10% or more could be confidently detected within individual containers.8,9
Data for the percent of original concentration remaining on each study day are presented in Table 1. The concentration of domperidone in Oral Mix suspensions remained above 93% of the initial concentration for up to 91 days with storage in 3 types of bottles (amber PVC, amber glass, and amber PET) at 2 different temperatures (25°C and 4°C) and in oral syringes at 25°C (Table 1).
Table 1 Domperidone Concentration and Percent Remaining (Mean ± Coefficient of Variation)* on Each Study Day and Calculation of Time to Achieve 90% Remaining with 95% Confidence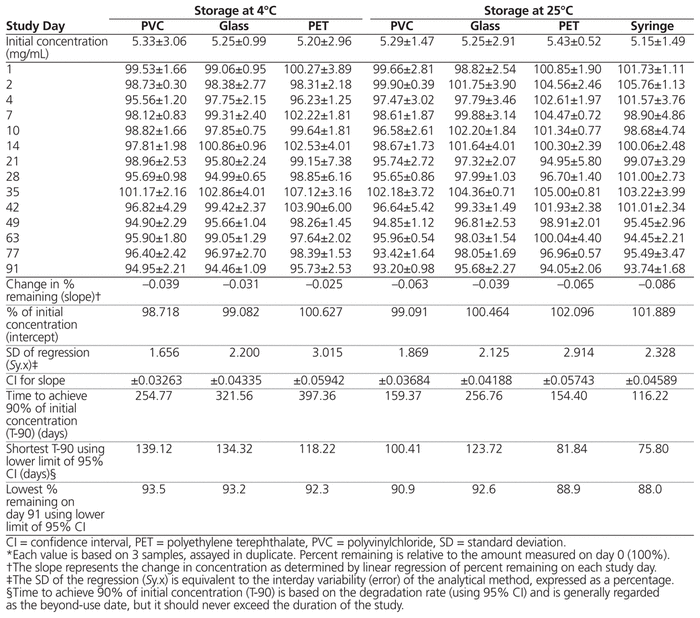
Interpretation of the study results with greatest confidence can be achieved through inspection of the amount remaining on day 91, determined with 95% confidence (see last row in Table 1). Inspection of these data reveals that there was a greater loss of concentration at room temperature and for suspensions stored in PET bottles. More specifically, for suspensions stored in PET bottles, there was an additional loss of 1% with storage at 4°C and an additional loss of almost 3.4% with storage at room temperature, relative to PVC and glass bottles. In PVC and glass bottles, more than 93% of domperidone concentration remained (with 95% confidence) with storage at 4°C; however, storage at room temperature resulted in an additional loss of 2.5% and 0.6% in PVC and glass bottles, respectively.
Analysis of variance showed no differences in percent remaining due to temperature (p = 0.29) but did show significant differences due to study day and container type (p < 0.001 for both). Similarly, multiple linear regression showed no differences in percent remaining due to temperature (p = 0.94) but did show significant differences due to study day and container type (p < 0.001 for both).
This study has demonstrated the stability of 5 mg/mL domperidone suspensions stored in amber PVC bottles, amber glass bottles, amber PET bottles, and clear polypropylene oral syringes. Suspensions retained at least 93.2% of the initial concentration for the entire 91-day study period. Even so, after calculation of the 95% confidence limits, we recommend that the BUD not exceed 75 days for suspensions stored at 4°C or 25°C.
The results of this study are very similar to those reported by Ensom and others.1 Re-analysis of those earlier data1 by a method identical with the method used in this study showed an estimated amount remaining on day 91 ranging from 81.4% to 94.5%, for suspensions with concentration of 1 mg/mL or 10 mg/mL stored in amber plastic bottles.
Ensom and others1 reported that some samples assayed higher than 100% during the study period, which was also observed in the current study. However, variation in the analytical method due to instrumentation or volume variation with dilution, along with the technique for sampling a suspension, is very important. In the current study, assay variability averaged less than 2.5%. Although the data for some of the containers indicated that domperidone would be stable for longer than 91 days, the BUD should never be extrapolated past the last day in a stability study. Furthermore, because some suspensions (those in PET bottles and oral syringes stored at room temperature) produced a calculated BUD, with 95% confidence, that was shorter than 91 days, and because there was no significant difference among the tested temperature–container combinations, we recommend the shortest BUD estimated to ensure confidence in the dose delivered on the 75th day after preparation.
It must be appreciated that stability studies are conducted in controlled environments. In real life, compounded suspensions will be stressed during use in hospitals or the home environment. For example, suspensions will be removed from the fridge on a daily basis to retrieve doses, and suspensions intended for refrigerated storage may be inadvertently left out of the fridge for long periods and at temperatures possibly well above the 25°C study temperature tested here. Furthermore, airspace will be created and will increase over time as the suspension is used up. Therefore, a decrease in concentration over time (as was observed with domperidone in this study, especially when using the 95% CI calculation) could result in delivery of a dose up to 10% less than the intended dose. Historically, BUDs have been tied to the time when a product reaches 90% of the label claim.10 However, we feel that this practice should be reviewed in light of the fact that stability studies are conducted in completely controlled laboratory environments. For drugs with a narrow therapeutic range, giving a dose that is 10% less than the intended dose could result in treatment failure. For this reason, we advocate the use of the 95% CI calculations, and our BUDs are based on those results, but never exceeding the duration of the study.
When a product is stored under a defined set of conditions, the observed concentration on each study day will be determined by the initial concentration, the degradation rate, the analytical variability, and the duration of storage. During a study, there will be some uncertainty associated with these observed concentrations because of error in the analytical method (accuracy and reproducibility). Therefore, daily concentrations reported in a stability study should be viewed as random estimates of the true concentration on that study day. As a result of the variability around the true concentration on any study day, the overall degradation rate from the study must be inferred from the data, and this degradation rate is best calculated using linear regression. The slope of this line is the degradation rate, and it has units of “percent per day” when the data are presented in terms of the percentage remaining.
This study had the power to detect differences in concentration of more than 3%. Neither analysis of variance nor multiple linear regression showed significant changes due to temperature; in comparisons within each container type, this translates into differences in the percent of initial concentration remaining on day 91 of less than 3%. Suspensions stored in all bottle types at room temperature or under refrigeration had differences in the average percentage remaining on day 91 of less than 2.6%, so these differences should also be regarded as nonsignificant and of no practical importance. However, at room temperature, the percent remaining in syringes relative to glass bottles differed by almost 5%. This is a statistically significant difference, and since the concentration on day 91 was determined to be less than 90% (with 95% confidence), this 5% difference is also a clinically important difference. Based on the shortest time to achieve 90% remaining (with 95% confidence), we recommend that the BUD for domperidone suspensions stored at room temperature in PET containers and oral syringes be no more than 75 days. This BUD provides assurance that suspensions stored at room temperature for less than 75 days will contain not less than 90% of the initial concentration.
Acknowledgement of analytical variability leads to the realization that the “best estimate” of the degradation rate from study data is just that: an estimate of the true degradation rate. Furthermore, there needs to be confidence that the true degradation rate is not faster than the best estimate. This is frequently ensured through use of a 95% CI.11–13
Assurance of the specificity of the analytical method is also important. The separation and detection of intact drug in the presence of degradation compounds must be demonstrated before the method can be considered stability-indicating. Furthermore, the accuracy and reproducibility of the assay in validation studies and during the stability study (on every assay day) provides the required confidence in the assay methodology, which is absolutely critical in any stability study.
The containers tested in this study included PVC bottles, which are no longer sold by Richards Packaging. However, we felt it was appropriate to test and report results for bottles of this type, using a previously obtained supply, because they may still be available and in use elsewhere in the world.
With storage at 4°C in various types of containers (PVC bottles, glass bottles, PET bottles, plastic syringes), more than 90% of the original concentration of domperidone remained after 91 days, with 95% confidence; however, the same did not hold true for suspensions of this drug stored at 25°C. Given that most pharmacies would prefer to store compounded domperidone suspensions at 25°C, for convenience, it is suggested that a shorter BUD of 75 days be assigned.
1 Ensom MHH, Decarie D, Hamilton DP. Stability of domperidone in extemporaneously compounded suspensions. J Inform Pharmacother. 2002; 8:100–4.
2 Trissel LA. Avoiding common flaws in stability and compatibility studies of injectable drugs. Am J Hosp Pharm. 1983;40(7):1159–60.
3 Trissel LA, Flora KP. Stability studies: five years later. Am J Hosp Pharm. 1988;45(7):1569–71.
4 Policy for publication of chemical stability study manuscripts. Can J Hosp Pharm. 1990;43(1):3.
5 Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, et al. Analytical methods validation: bioavailability, bioequivalence, and pharmacokinetic studies. J Pharm Sci. 1992;81(3):309–12.
6 Oral Mix™ (flavored suspending vehicle) [safety data sheet]. Medisca; [revised 2017 Sep; cited 2018 May 14]. Available from: https://www.medisca.com/NDC_SPECS/MUS/2512/MSDS/2512.pdf
7 PreciseDose dispenser™ w/tip cap. Product no. 8147 [specification sheet]. Medisca [cited 2018 May 14]. Available from: https://www.medisca.com/NDC_SPECS/MUS/8147/Downloads/8147%20Medisca%20Spec%20Sheet.pdf
8 Frieman JA, Chalmers TC, Smith H Jr, Kuebler RR. The importance of beta, the type II error and sample size in the design and interpretation of the randomized control trial—survey of 71 negative trials. N Engl J Med. 1978; 299(13):690–4.
9 Stolley PD, Strom BL. Sample size calculations for clinical pharmacology studies. Clin Pharmacol Ther. 1986;39(5):489–90.
10 General chapter <797>: pharmaceutical compounding—sterile preparations. In: USP compounding compendium. Rockville (MD): US Pharmacopeial Convention Inc; 2018. p. 39–82.
11 Carstrensen JT, Franchini M, Ertel K. Statistical approaches to stability protocol design. J Pharm Sci. 1992;81(3):303–8.
12 Shao J, Chow SC. Statistical inference in stability analysis. Biometrics. 1994; 50(3):753–63.
13 Magari RT. Uncertainty of measurement and error in stability studies. J Pharm Biomed Anal. 2007;45(1):171–5.
Funding: This study was funded by an unrestricted educational grant from Medisca Inc, Saint-Laurent, Quebec, along with the Departments of Pharmacy at Sunnybrook Health Sciences Centre and The Hospital For Sick Children, Toronto, Ontario. Medisca was not involved in the design or conduct of the study; the collection, management, or interpretation of the data; or the preparation, review, or approval of the manuscript. ( Return to Text )
Competing interests: Other than study funding, no competing interests were declared. ( Return to Text )
The authors thank Pacita Sales for assistance with the initial physical study, design of the study suspension, and preparation of the study suspensions.
Canadian Journal of Hospital Pharmacy, VOLUME 71, NUMBER 3, May-June 2018