Stephanie Gautron, Jason Wentzell, Salmaan Kanji, Tiffany Nguyen, Daniel M. Kobewka, Erika MacDonaldABSTRACT
Background
The Protecting Canadians from Unsafe Drugs Act will eventually require institutions to report all serious adverse drug reactions (ADRs), although the proposed regulations do not yet define what will need to be reported and by whom. Knowledge about the occurrence of serious ADRs in the hospital setting is needed to optimize the effectiveness of reporting and to determine the potential implications of mandatory reporting.
Objectives
To quantify and characterize suspected serious ADRs in patients admitted to a general medicine service, to assess the likelihood of causality, and to determine inter-rater agreement for identification of ADRs and assessment of their likelihood.
Methods
This prospective observational study involved 60 consecutive patients admitted to a general medicine service at a tertiary care teaching centre starting on March 28, 2016. The primary outcome was the number of serious ADRs, defined by Health Canada as ADRs that result in hospital admission, congenital malformation, persistent or significant disability or incapacity, or death; that are life-threatening; or that require significant intervention to prevent one of these outcomes. Medical records were reviewed independently by pairs of pharmacists for serious ADRs, and the likelihood of causality was assessed using the World Health Organization–Uppsala Monitoring Centre system. Inter-rater agreement was calculated using the kappa score, and disagreements were resolved by discussion and consensus.
Results
Twenty-three serious ADRs occurred in the sample of 60 patients. The proportion of patients experiencing a serious ADR that contributed to the original hospital admission was 19/60 (32%, 95% confidence interval [CI] 20%–43%), and 4 patients (7%, 95% CI 0%–13%) experienced a serious ADR during their hospital stay. Inter-rater agreement for occurrence of serious ADRs was moderate (kappa 0.58, 95% CI 0.35–0.76).
Conclusion
Reportable serious ADRs were common among patients admitted to a general medicine service. Canadian hospitals would face difficulties reporting all serious ADRs because of the frequency of their occurrence and the subjectivity of their identification.
KEYWORDS: adverse drug reactions, postmarket surveillance, adverse drug reaction reporting, hospital pharmacy
RÉSUMÉ
Contexte
La Loi visant à protéger les Canadiens contre les drogues dangereuses obligera éventuellement les établissements à déclarer tout cas de réactions indésirables graves aux médicaments (RIM), quoique les règlements proposés n’indiquent pas encore ce qui devra être déclaré et par qui. Des données sur la survenue de RIM graves en milieu hospitalier sont nécessaires pour optimiser l’efficacité de la déclaration et pour déterminer les implications potentielles d’une déclaration obligatoire.
Objectifs
Quantifier les RIM graves soupçonnées chez les patients admis à un service de médecine générale et en offrir un portrait, évaluer la probabilité d’une relation de causalité et déterminer l’accord interévaluateurs pour le repérage des RIM et l’évaluation de leur probabilité.
Méthodes
La présente étude observationnelle prospective comptait 60 patients admis consécutivement à partir du 28 mars 2016 à un service de médecine générale d’un centre hospitalier universitaire de soins tertiaires. Le principal paramètre d’évaluation était le nombre de RIM graves, définies par Santé Canada comme des RIM qui mènent à une hospitalisation, à une malformation congénitale, à une invalidité ou à une incapacité persistante ou importante; qui mettent la vie en danger ou entraînent la mort; ou qui nécessitent une intervention significative pour prévenir l’un de ces résultats. Les dossiers médicaux ont été examinés indépendamment par des paires de pharmaciens à la recherche de RIM graves et la probabilité d’une causalité a été évaluée à l’aide du système du Centre de pharmacovigilance d’Uppsala de l’Organisation mondiale de la Santé. L’accord interévaluateurs a été mesuré à l’aide du coefficient kappa et les désaccords ont été résolus par la discussion et l’atteinte d’un consensus.
Résultats
Vingt-trois RIM graves sont survenues dans l’échantillon composé de 60 patients. La proportion de patients ayant subi une RIM grave qui a contribué à l’hospitalisation initiale était 19/60 (32 %, intervalle de confiance [IC] de 95 % de 20 %–43 %); de plus, 4 patients (7 %, IC de 95 % de 0 %–13 %) avaient subi une RIM grave au cours de leur séjour à l’hôpital. L’accord interévaluateurs sur la survenue de RIM graves était modéré (kappa = 0,58, IC de 95 % de 0,35–0,76).
Conclusion
Les RIM graves à déclaration obligatoire étaient courantes chez les patients admis à un service de médecine générale. Les hôpitaux canadiens auraient de la difficulté à déclarer tous les cas de RIM graves à cause de leur fréquence et de la subjectivité de leur repérage.
MOTS CLÉS: réactions indésirables aux médicaments, pharmacovigilance, déclaration des réactions indésirables aux médicaments, pharmacie hospitalière
Health Canada relies on spontaneous reporting of adverse drug reactions (ADRs) to optimize the postmarket safety of medications. Previously unknown ADRs are often identified in clinical practice, and pharmacovigilance centres rely primarily on voluntary reporting of ADRs by health professionals.1 Reporting is particularly important for ADRs that are rare or that occur only after long-term use, as these types of ADR are not likely to be identified in premarket clinical trials.1 Important safety signals arising from spontaneous ADR reports have led to regulatory actions, including withdrawal of drugs from the market, labelling changes, public alerts, and notices sent to health professionals.2 For example, the prescription drug cisapride, indicated for the treatment of refractory gastroparesis, intestinal pseudo-obstruction, and gastroesophageal reflux disease, was approved for the Canadian market in 1991.3,4 Health Canada subsequently received 44 spontaneous ADR reports of cardiac rhythm abnormalities in patients taking cisapride, including 10 reports of death associated with the use of this drug. Likewise, the US Food and Drug Administration received 341 ADR reports of cardiac rhythm abnormalities, including 80 reports of death associated with its use. These spontaneous ADR reports led to changes in the product monograph, safety warnings, and the eventual withdrawal of cisapride from the Canadian market (in the year 2000).4
Currently, the reporting of ADRs to Health Canada by health professionals is voluntary. Health Canada’s ADR reporting guideline5 states that “any suspected” ADR should be reported, especially those that are “unexpected” (not consistent with product information or labelling), regardless of their severity; those that are serious, whether expected or not; and those related to a health product that has been on the market for less than 5 years.
Adverse reactions are defined in the Health Canada guideline as “noxious and unintended effects to health products”.5 The guideline notes that adverse reaction reports are most typically “only suspected associations” and that a health professional does not have to be certain that the reaction was due to a drug (or other health product) in order to report the reaction.5 A serious adverse reaction is defined as “one which requires hospitalization or prolongation of existing hospitalization, causes congenital malformation, results in persistent or significant disability or incapacity, is life-threatening, or results in death. Adverse reactions that require significant medical intervention to prevent one of these outcomes are also considered to be serious.”5
The Protecting Canadians from Unsafe Drugs Act, which was enacted in November 2014, aims to improve Health Canada’s ability to collect postmarket safety information. As part of this act, also known as Vanessa’s Law, institutions will be required to report all serious ADRs.6 However, this requirement is not yet being enforced, because supporting regulations are not yet available.7
Subjectivity in the identification of serious ADRs will make it difficult to enforce mandatory reporting by health professionals. Signs and symptoms of ADRs can be nonspecific, and it is therefore often difficult for clinicians to differentiate an ADR from a current illness.8 Furthermore, many clinicians report lack of time as a barrier to ADR reporting.9 Additionally, reporting reactions that are well established as being associated with a particular drug is likely not an optimal use of scarce resources. For example, there is a multitude of evidence that warfarin causes bleeding and that benzodiazepines cause sedation and delirium. Reporting of suspected serious ADRs like these, which are well-known side effects of medications that occur with high frequency, would require substantial resources and would be unlikely to improve knowledge of a medication’s safety.
The foregoing considerations indicate that reporting all suspected serious ADRs has uncertain benefit and, furthermore, that institutions in Canada may face difficulties meeting the requirements of the Protecting Canadians from Unsafe Drugs Act. We therefore conducted a prospective observational study of patients admitted to a general medicine service at a tertiary care teaching hospital to determine the type and frequency of suspected serious ADRs, as well as inter-rater agreement in these determinations. We also characterized the ADRs to inform the operationalization of the Protecting Canadians from Unsafe Drugs Act for hospitals.
This study had the following objectives:
to quantify the number of suspected serious ADRs in patients admitted to a general medicine service
to characterize the suspected serious ADRs in patients admitted to this general medicine service
to assess the likelihood of causality of the suspected serious ADRs in patients admitted to this general medicine service
to determine inter-rater agreement for identification of suspected serious ADRs and assessment of their likelihood.
This prospective observational study was based on data from patients’ health records. Ethics approval was obtained from the Ottawa Health Science Network Research Ethics Board, which waived the need for informed consent.
The study took place at a 1122-bed tertiary care teaching centre in Ontario, Canada. Consecutive patients admitted through the emergency department to the general medicine service at the largest campus of this institution (over a period of 8 consecutive days starting on March 28, 2016) were eligible for inclusion. Patients were excluded if the investigators could not access their paper charts. Patients’ data were censored if the hospital stay extended beyond 28 days.
Consecutive patients were considered for inclusion until the prespecified sample size of 60 was reached. The sample size of 60 was chosen because chart review for this number of patients was feasible, given resources available at the time of the study. Also, it allowed for adequate precision for estimates of proportions: the 95% confidence interval (CI) would be precise to ±13% at maximum variance (i.e., a proportion of 50%).
The World Health Organization–Uppsala Monitoring Centre system for standardized case causality assessment (referred to hereafter as the WHO-UMC system) was chosen as the method for determining the likelihood of causality of each ADR. The WHO-UMC system classifies ADRs into 6 broad categories: certain, probable/likely, possible, unlikely, conditional/unclassified, and unassessable/unclassifiable.10 To classify the likelihood of causality, the assessor considers the event, the plausibility of the time relationship, the response to withdrawal (if applicable), and other possible contributing factors. Complete definitions for the WHO-UMC causality categories are presented in Appendix 1.
A single investigator, a pharmacy resident licensed as a pharmacist (S.G.), collected data from the paper and electronic health records of all included patients. For each patient, a second pharmacist (from a pool of 4 pharmacists with hospital residency training who were practising in a range of clinical areas, all with 7 or more years of experience in inpatient care [J.W., S.K., T.N., E.M.]) independently reviewed and assessed the health records for the occurrence of suspected serious ADRs, and assessed the likelihood of causality of any suspected serious ADRs according to the WHO-UMC system. Specifically, for each patient, the 2 pharmacists independently reviewed the admission diagnoses (primary and contributing) from the admission consult, the best possible medication history, the admitting team’s daily progress notes, documentation from consulting services, medications ordered in hospital, and discharge summaries to identify suspected serious ADRs and to assess their likelihood. Laboratory measures, vital signs, and the medication administration record were reviewed when either of the pharmacists deemed this information to be relevant to the assessment of a suspected serious ADR. This chart review was intended to reflect how a pharmacist would review the chart during the course of usual patient care. The 2 pharmacists independently assessed whether a primary or contributing admission diagnosis should be considered to represent a suspected serious ADR. Additionally, the 2 pharmacists independently assessed whether a suspected serious ADR occurred during the hospital stay. For each suspected ADR, the 2 pharmacists used the WHO-UMC system to independently assess the likelihood that a particular drug caused the reaction. Disagreements were resolved by discussion and consensus. When deemed necessary by either of the 2 pharmacists, drug product monographs, accessed from Health Canada’s Drug Product Database, were reviewed to help make the assessment of likelihood. Other references, such as Lexi-Drugs, Micromedex, and MedEffect Canada databases, were also accessed at the discretion of the pharmacists.
The pharmacists did not communicate with a patient’s health care team unless they felt that they had identified a serious ADR of which the team was unaware and that warranted intervention. ADRs identified during the course of the study were reported to Health Canada at the discretion of the team pharmacist (not the study investigators), in accordance with Health Canada’s current guidance and current practice at the study institution.
The primary outcome was the number of suspected serious ADRs contributing to the reason for hospital admission or occurring during the hospital stay.
Secondary outcomes were the number of suspected serious ADRs contributing to the reason for hospital admission; the number of suspected serious ADRs occurring during the hospital stay; the proportions of patients experiencing suspected serious ADRs (overall, upon admission, and during the hospital stay); the mean number of suspected serious ADRs per patient; the proportion of suspected serious ADRs that were unexpected (where, for the purposes of this study, an “unexpected” reaction was one not listed in the product monograph), the proportion that were caused by a “new” drug (one that had been on the market for less than 5 years), and the proportion that met both of these criteria; the proportion of suspected serious ADRs that were considered to be possible, likely, or certain according to the WHO-UMC system; and inter-rater reliability for the identification of a suspected serious ADR and for the classification of likelihood of a suspected serious ADR.
Data are reported as frequencies and proportions, with 95% CIs as appropriate. A kappa score was calculated to quantify inter-rater agreement for the identification of suspected serious ADRs and their likelihood. Excel (Microsoft Corporation, Redmond, Washington) and SAS version 9.4 statistical software (SAS Institute Inc, Cary, North Carolina) were used for all statistical analyses.
A total of 67 consecutive, potentially eligible patients were admitted to the general medicine service between March 28 and April 4, 2016, but 7 patients were excluded from analysis because their paper charts were inaccessible. The mean age of the 60 patients included in the analysis was 67 (range 19–95) years, and the mean number of medications at the time of admission, as recorded in the patients’ admission medication history was 11 (range 0–25). Thirty-two (53%) of the patients were men.
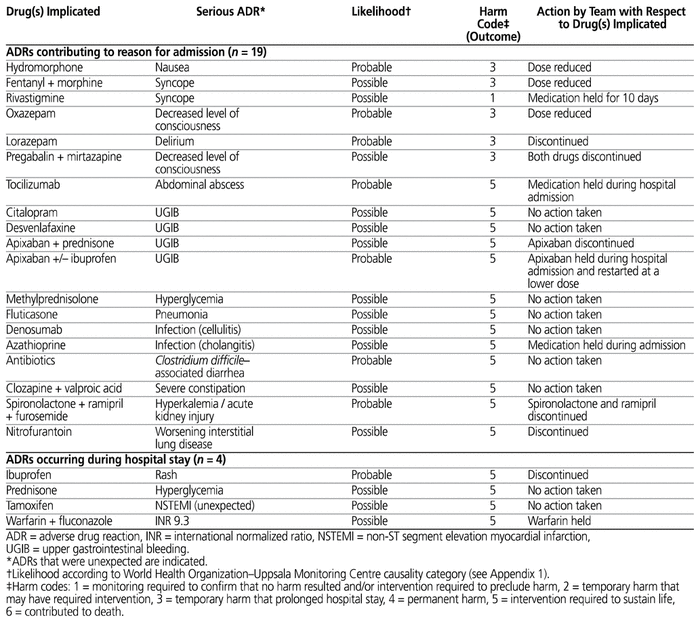
A total of 23 serious ADRs were identified in 21 of the 60 patients. For each ADR, the drug (or drugs) implicated and the associated reactions, outcomes, and actions taken with regard to the implicated drugs are described in Table 1.
Table 1 Characteristics of Suspected Serious Adverse Drug Reactions
Nineteen of the 23 serious ADRs contributed to hospital admission for a total of 19 of the 60 patients (32%, 95% CI 20%–43%). Four of the 23 serious ADRs occurred during the hospital stay for a total of 4 patients (7%, 95% CI 0%–13%). Two of the patients each experienced 2 serious ADRs; as such, a total of 21 of the 60 patients (35%, 95% CI 23%–47%) experienced a suspected serious ADR contributing to the reason for hospital admission and/or during the hospital stay. The mean number of suspected serious ADRs per patient was 0.4.
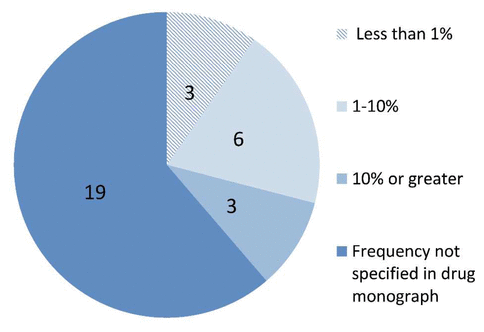
The proportion of suspected serious ADRs caused by a new drug (marketed for less than 5 years) was 2/23 (9%). Only one (4%) of the 23 suspected serious ADRs was considered unexpected (not described in the product monograph). None of the suspected ADRs met both of these criteria. Figure 1 depicts the expected frequency of occurrence of an ADR, as stated in the product monographs for the 31 drug–ADR pairs identified in this study (as listed in Table 1). According to the WHO-UMC system, 15 (65%) of the 23 serious ADRs were considered possible, 8 (35%) were considered probable, and none were considered certain.
|
|
||
|
Figure 1 Frequency of occurrence of adverse drug reactions (ADRs) as reported in monograplns for the 31 drug–ADR pairs listed in Table 1. For the purpose of this figure, an ADR for which 2 drugs were implicated (e.g., syncope in a patient taking fentanyl + morphine) generated 2 drug–ADR pairs. |
||
In 12 instances, one of the pharmacists identified a serious ADR that the other pharmacist did not identify. After discussion and consensus, 9 of these events were included as ADRs for the purposes of the analysis, and 3 were excluded. The kappa score for inter-rater agreement on identification of ADRs was 0.58 (95% CI 0.35–0.76), indicating a moderate level of inter-rater agreement. There was disagreement in the assessment of likelihood for 4 of the 23 ADRs. In each of these 4 instances, one of the pharmacists considered the reaction to be “probable” and the other pharmacist considered it to be “possible” (i.e., for these 4 ADRs, neither of the pharmacists assessed the likelihood as “certain”). The kappa score for inter-rater agreement for likelihood of a suspected serious ADR was also moderate, at 0.62 (95% CI 0.28–0.96).
One-third of general medicine patients admitted to a Canadian tertiary care teaching centre experienced a suspected serious ADR contributing to the reason for hospital admission or occurring during their hospital stay. Only a small proportion of these ADRs were unexpected (4%) or caused by drugs that were newly marketed (9%), with none of the ADRs meeting both of these criteria. None of the suspected serious ADRs were classified by the assessing pharmacists as “certain” according to the WHO-UMC causality classification system, and only 35% were classified as “probable”, whereas the majority (65%) were classified only as “possible”. By definition, a possible reaction is one that “could also be explained by disease or other drugs”.10 Classification of the majority of suspected serious ADRs as possible (rather than probable or certain) reinforces the inherent ambiguity of the identification of ADRs. In many instances, no changes were made to the patient’s regimen for the drugs implicated in the adverse reactions (see Table 1). This outcome is not surprising, given that “possible reactions” will, by definition, often have other potential explanations.
Although only one of the ADRs in this study was classified as unexpected, the definition of “unexpected” is another point of ambiguity. We elected to classify a reaction as unexpected only if it was not listed as a possible adverse effect in the product monograph. In other words, for the purposes of our study, any reaction mentioned in the product monograph would not have been classified as unexpected, regardless of rarity and regardless of whether the reaction was detected only in postmarketing case reports. If regulations for the mandatory reporting of serious ADRs were to require the reporting of unexpected reactions only (as opposed to all serious ADRs), Health Canada would need to carefully consider the definition of “unexpected”. For example, the definition could include reactions that are listed in the project monograph but that occur only rarely or for which causality has not been established.
The frequency of occurrence of serious ADRs as reported in other studies varies substantially, likely because of differences in the populations studied, differences in methodology, a lack of standard definitions for “ADR” and “adverse drug event”, and subjectivity in outcome assessment. Several previous studies have reported on the occurrence of adverse drug events and/or ADRs in Canadian hospitals. Samoy and others11 conducted a prospective observational study of 565 adult patients admitted to the internal medicine service of a Canadian hospital in which the frequency of drug-related hospital admissions was 24.1%. However, ADRs were just 1 of 8 categories of adverse drug events potentially leading to admission, and only adverse drug events relating to the chief complaint on admission were considered. Forster and others12 examined the occurrence of adverse events in a random sample of 502 patients admitted with nonpsychiatric illness to a single institution. The incidence of any adverse event was 12.7%, and 50% of these adverse events were deemed to be due to a drug (i.e., adverse drug events). The results of the current study are not consistent with these previous results, for several possible reasons. For example, the study methodologies were different. In addition, in the earlier studies, occurrence of an ADR had to have been documented in the chart at the time of patient care in order for the event to be counted as an outcome, whereas in our study a pharmacist critically reviewed the patient health record in an attempt to identify suspected ADRs.
Sikdar and others13 found a prevalence of adverse drug events of only 2.4% in a sample of 1458 patients presenting to the emergency departments at 2 tertiary care hospitals in St John’s, Newfoundland and Labrador. This estimate was much lower than what is reported here for ADRs, especially considering that the definition of “adverse drug event” is broader than (and indeed encompasses) the definition of “ADR”. The difference in outcome estimates may be due to population differences, as the St John’s study was not restricted to internal medicine and included patients who were not admitted to hospital from the emergency department. Additionally, the chart reviews in the St John’s study were conducted retrospectively,13 whereas our study was prospective.
The frequency of occurrence of ADRs has also been reported in meta-analyses. A 1998 meta-analysis of 39 prospective studies conducted in hospitals in the United States reported an incidence of serious ADRs causing hospital admission or occurring in hospital of 6.7% (95% CI 5.2%–8.2%).14 The lower frequency in that study, as compared to the study reported here, may be explained by the exclusion of serious ADRs that were classified as “possible”, defined as those that followed a “reasonable temporal sequence and for which the ADR is a known response to the drug, although the response may also be explained by the patient’s clinical state.”14 In our study, 8 (13%) of the 60 patients experienced an ADR that was considered probable (i.e., when those with ADRs considered as “possible” were excluded). A more recent meta-analysis of 22 prospective studies, published in 2012, found that 16.9% (95% CI 13.6%–20.2%) of patients experienced an ADR during their hospitalization.15 All suspected ADRs were included in this analysis, not only those that were serious. The validity of this pooled estimate is questionable, however, because significant statistical heterogeneity was observed (I2 = 99%). The authors stated that differing methodologies represented the most important contributor to heterogeneity across the included studies.15
The results of the current study suggest that indiscriminate reporting of all suspected serious ADRs would unnecessarily burden clinicians. Assuming that preparation of an ADR form for submission to Health Canada takes 10–20 min per ADR, it would take 5–10 h/week to submit reports for all suspected serious ADRs occurring on the general medicine service at a single campus of our institution. This time estimate is anecdotal, based on our own and our colleagues’ experience reporting ADRs. The majority of ADRs identified in this sample of 60 patients were well-known side effects, and the reporting of these established ADRs would be unlikely to improve knowledge about the safety of these medications, despite the substantial investment of time required.
As described in a public consultation on the mandatory reporting of serious ADRs held in summer 2017, proposed changes to the regulations have not yet been finalized.16 Which health care institutions should report ADRs, what types of serious ADRs will be reported, what information should be included in an ADR report, and the expected timelines for reporting have not yet been defined. Clear guidance for health professionals that prioritizes reporting of those ADRs most likely to increase medication safety knowledge could increase the effectiveness and feasibility of mandatory reporting.
Subjectivity in ADR identification will make it difficult to execute and enforce mandatory reporting by health professionals. In our study, pairs of pharmacists identified and assessed the likelihood of causality for all suspected serious ADRs that were identified, with moderate inter-rater agreement. The signs and symptoms of ADRs can be nonspecific and indistinguishable from symptoms of the underlying disease; therefore, it is often difficult for clinicians to differentiate an ADR from a current illness.8
Our study had several limitations. The results may not reflect the incidence of suspected serious ADRs outside of the general medicine service at the study institution or at other institutions. The study was conducted over a single 8-day period and involved a relatively small number of patients at a single institution. Identification of ADRs was limited by the information available in the patients’ health records. Discussions with patients, caregivers, and the medical team might have led to identification of additional suspected ADRs or a different classification of likelihood, but such discussions were not feasible. The definition of “ADR” used in this study was the one provided in Health Canada’s guideline on adverse reaction reporting for health professionals.5 Definitions of “ADR” and “adverse drug event” are not consistently reported in the available literature, which makes it challenging to compare and synthesize the results of different studies. Furthermore, there is currently no universally accepted method for assessing the causality of suspected ADRs.8 The WHO-UMC system was chosen as the method of causality assessment for this study because it is a practical tool for determining the likelihood of causality of an ADR that is based on spontaneous ADR data from around the world, as received and analyzed by the WHO’s Uppsala Monitoring Centre. Although the Naranjo algorithm is often used in research, it was not selected as a method of causality assessment in this study because it is less practical for application in practice, for several reasons; for example, it asks whether the reaction reappeared with the administration of a placebo, and it considers blood concentrations of the drug in question, information that is often not available when likelihood is assessed in practice.17
Suspected serious ADRs were identified in about one-third of patients admitted to a general medicine service at a tertiary care teaching centre. Institutions in Canada would likely face difficulties in reporting all suspected serious ADRs because of the frequency of their occurrence, subjectivity in the assessment of occurrence of ADRs and their likelihood, and the implications for health professionals’ workload. The majority of ADRs identified were well-known side effects, and reporting them would be unlikely to improve overall knowledge relating to medication safety.
The provision of clear guidance for health professionals with respect to the identification of reportable suspected serious ADRs and assessment of the likelihood of causality could minimize false safety signals and improve medication safety.
Reproduced, with permission of the publisher, from: The Use of the WHO-UMC System for Standardised Case Causality Assessment. Uppsala (Sweden): Uppsala Monitoring Centre; 2018. Available from: https://www.who-umc.org/media/164200/who-umc-causality-assessment_new-logo.pdf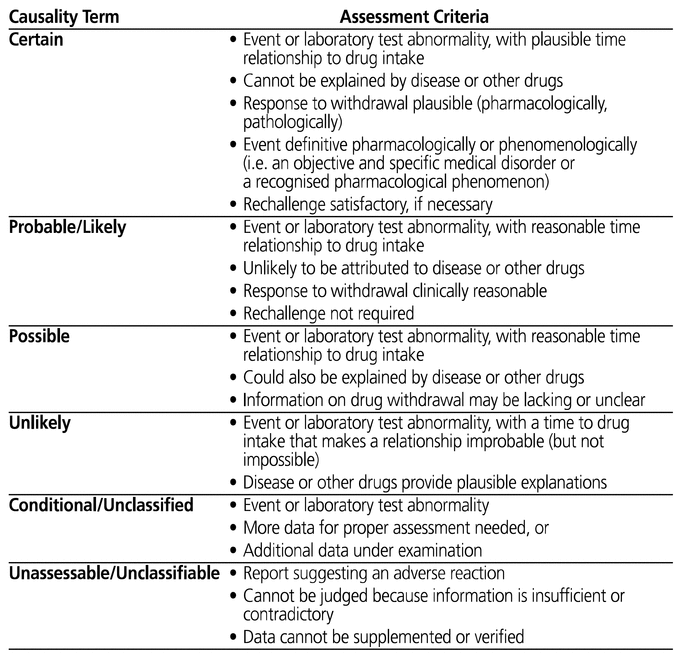
1 Dal Pan GJ, Lindquist M, Gelperin K. Postmarketing spontaneous pharmacovigilance reporting systems. In: Strom BL, editor. Textbook of pharmacoepidemiology. 2nd ed. Hoboken (NJ): John Wiley & Sons, Ltd; 2013. p. 101–17.
2 Wiktorowicz ME, Lexchin J, Moscou K, Silversides A, Eggertson L. Keeping an eye on prescription drugs, keeping Canadians safe. Active monitoring systems for drug safety and effectiveness in Canada and internationally. Toronto (ON): Health Council of Canada; 2010 [cited 2015 Nov 4]. Available from: http://publications.gc.ca/collections/collection_2011/ccs-hcc/H174-21-2010-eng.pdf
3 Lexchin J. How safe are new drugs? Market withdrawal of drugs approved in Canada between 1990 and 2009. Open Med. 2014;8(1):e14–9.

4 Peterson RG. Recalls and safety alerts: Prepulsid (cisapride). Ottawa (ON): Government of Canada; 2000 [cited 2017 Jun 6]. Available from: http://healthycanadians.gc.ca/recall-alert-rappel-avis/hc-sc/2000/14309a-eng.php?_ga=2.128078860.1095885074.1496795372-1085454810.1496795372
5 Adverse reaction reporting and health product safety information - guide for health professionals. Ottawa (ON): Health Canada; 2011 [cited 2015 Nov 6]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/reports-publications/medeffect-canada/adverse-reaction-reporting-health-product-safety-information-guide-health-professionals-health-canada-2011.html
6 Protecting Canadians from Unsafe Drugs Act (Vanessa’s Law) amendments to the Food and Drugs Act (Bill C-17). Ottawa (ON): Health Canada; 2015 [modified 2016 Jul 29; cited 2015 Nov 4]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/legislation-guidelines/protecting-canadians-unsafe-drugs-act-vanessa-law-amendments-food-drugs-act.html
7 Norris S, Tiedemann M. Legislative summary of Bill C-17: an act to amend the Food and Drugs Act. Ottawa (ON): Parliament of Canada; 2014 [cited 2015 Nov 4]. Available from: www.parl.gc.ca/About/Parliament/LegislativeSummaries/bills_ls.asp?source=library_prb&ls=C17&Parl=41&Ses=2&Language=E&Mode=1#a9
8 Rohan CH, Kinage PJ, Gaikwad NN. Causality assessment in pharmacovigilance: a step towards quality care. Sch J Appl Med Sci. 2013;1(5):386–92.
9 Peterson LN, Peterson R, Ho K, Olatunbosun T. Barriers to Canadian physicians reporting of adverse drug reactions. Vancouver (BC): University of British Columbia; 2009 [cited 2016 March 21]. Available from: http://dx.doi.org/10.14288/1.0055435
10 The use of the WHO-UMC system for standardised case causality assessment. Uppsala (Sweden): Uppsala Monitoring Centre; 2018 [cited 2018 Oct 3]. Available from: https://www.who-umc.org/media/164200/who-umc-causality-assessment_new-logo.pdf
11 Samoy LJ, Zed PJ, Wilbur K, Balen RM, Abu-Laban RB, Roberts M. Drug-related hospitalizations in a tertiary care internal medicine service of a Canadian hospital: a prospective study. Pharmacotherapy. 2006; 26(11):1578–86.
12 Forster AJ, Asmis TR, Clark HD, Al Saied G, Code CC, Caughey SC, et al. Ottawa Hospital Patient Safety Study: incidence and timing of adverse events in patients admitted to a Canadian teaching hospital. CMAJ. 2004;170(8):1235–40.
13 Sikdar KC, Alaghehbandan R, MacDonald D, Barrett B, Collins KD, Donnan J, et al. Adverse drug events in adult patients leading to emergency department visits. Ann Pharmacother. 2010;44(4):641–9.
14 Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998; 279(15):1200–5.
15 Miguel A, Azevedo LF, Araújo M, Pereira AC. Frequency of adverse drug reactions in hospitalized patients: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2012;21(11):1139–54.
16 Consultation: mandatory reporting of serious adverse drug reactions and medical device incidents. Ottawa (ON): Government of Canada; [updated 2017 Aug 15; cited 2018 Feb 5]. Available from: https://www.canada.ca/en/health-canada/programs/consultation-reporting-serious-adverse-drug-reactions-medical-device-incidents.html
17 Kane-Gill SL, Forsberg EA, Verrico MM, Handler SM. Comparison of three pharmacovigilance algorithms in the ICU setting: a retrospective and prospective evaluation of ADRs. Drug Saf. 2012;3(8):645–53.
Competing interests: None declared. ( Return to Text )
Address correspondence to: Erika MacDonald, The Ottawa Hospital, Civic Campus, 1053 Carling Avenue, Ottawa ON K1Y 1J8, e-mail:erimacdonald@toh.on.ca
Funding: None received. ( Return to Text )
Canadian Journal of Hospital Pharmacy, VOLUME 71, NUMBER 5, September-October 2018