Jennifer Fenna, Kathy Watkins, Micheal Guirguis
Biologics, a class of large, complex drugs that are derived from living organisms rather than chemical synthesis,1,2 represent an emerging area of drug therapy. Biologic drugs are important treatment options often used in chronic diseases and cancer. From a health care system perspective, they are a major contributor to rising drug expenditures.3,4 However, as patents for branded products expire, there is an opportunity to develop biosimilar drugs in Canada.1,5 Biosimilars are highly similar versions of biologics already authorized for market,5 which are comparably efficacious and safe, but potentially less expensive.6
As the market for biosimilars grows, their inclusion in hospital formularies can benefit the health care system and improve quality of care by increasing patients’ access to treatment options and reducing expenditures.3 However, biosimilars have class- and product-specific characteristics that differ from those of traditional small-molecule drugs,7 and a formulary evaluation to determine efficacy, safety, and cost-effectiveness requires additional considerations. Drugs and therapeutics committees (DTCs) play a critical role in evaluating biosimilars and improving their uptake in acute care. Ultimately, a DTC serves to establish and maintain a formulary that best meets the needs of prescribers, patients, and the health care organization.8
The purpose of this article is to describe one provincial health authority’s practice in adding biosimilars to its acute care formulary. A summary of the available literature on considerations when adding biosimilars to formulary is provided. This information is then applied to the experience of the authors’ health authority, to identify the strengths and challenges of its formulary review and decision process for biosimilar drugs. The article is intended to educate Canadian hospital pharmacists about formulary and practice management tools and principles, as well as educational strategies, to support the safe and effective use of biosimilars.
In order to be marketed, a biosimilar must be demonstrated to be sufficiently similar to the reference product, meaning there is no clinically meaningful difference in safety, purity, or efficacy.1,9,10 In Canada, biosimilars are approved via an abbreviated pathway. Using a totality-of-evidence approach, stepwise development begins with structural and functional studies and continues with human clinical studies.2 Relative to the originator biologics, biosimilars require fewer patients to be studied, and less clinical data to support their efficacy and safety, but must have more analytical information (e.g., structure and function).11 Additionally, biosimilars may be authorized for use for more than one indication (through extrapolation of therapeutic similarity from one indication to another), even if clinical studies have not been conducted for each indication.1
Biosimilars are not the same as generic versions of a drug,2 in that they are highly similar to, but not exact copies of, the originator product.12 Traditional small-molecule generics can be chemically synthesized and are identical with their respective reference products.12 In contrast, biologics are large proteins developed from living sources (e.g., bacteria), with manufacturing steps that are numerous and complex.12 As such, there is potential for differences among batches of the same biologic.1,11 Because the manufacturing process for any biologic is proprietary, it is impossible for a competitor to replicate all aspects of it.1
The strong similarity between a biosimilar and its corresponding biologic has implications for the types of formulary tools that can be used for evaluation.12,13 The concepts of substitution, inter-changeability, and switching must be understood in this regard (Table 1). Substitution is “the act of dispensing one product in place of another.”1 Interchangeability occurs when one drug can be substituted for another at the time of dispensing, with the substituted product being so like its originator that it is expected to achieve the same clinical effect in any given patient.1,5,13 In Canada, interchangeability decisions are made at the provincial/territorial level1,13; there are also some differences in how provinces define these concepts.5 Therapeutic interchange, also known as therapeutic substitution,1,13 is a formulary management tool that allows substitution of a different medication from the same class of drugs with similar therapeutic efficacy and safety.1,12 Therapeutic interchange is done at the pharmacy level and involves collaboration between the prescriber and the pharmacist.13,14 Therapeutic interchanges done by hospital pharmacists are based on a medical directive from the organization’s DTC or from an individual physician.1 This type of interchange differs from switching to a biosimilar, which involves changing a specific patient’s medication after therapy with a biologic has already been established for that patient.1 Decisions about switching are generally made by individual patients and their practitioners, on a case-by-case basis.1,13 At this time, Canadian guidance does not support the application to biosimilars of routine drug interchange or substitution practices that are commonly used for generic drugs.1,5,10
Table 1 Comparison of Generic and Biosimilar Drugs with Corresponding Brand
Key elements of formulary review of biosimilars include evaluation of clinical parameters (indications, clinical data, immunogenicity), product characteristics (nomenclature, supply management, packaging and labelling), and institutional considerations (substitution, pharmacovigilance, costs and reimbursement, patient and provider education, tracking and information system implications).3 Griffith and others11 developed a checklist of considerations to be taken into account when evaluating biosimilars for formulary inclusion. Although this checklist is based on the US health system perspective, the process can be applied to Canadian settings to ensure that all important elements are considered in formulary review of a biosimilar.
Little has been published on Canadian efforts in this area, although experiences from other parts of the world show that uptake has not reached its full potential, and many barriers exist to inclusion of biosimilars on formulary.11 Factors that limit the uptake of biosimilar products include limited prescriber awareness, lack of interchangeability between biosimilars and originator products, a need for more pharmacovigilance data to supplement clinical trial data, and differences in value-added elements such as manufacturer support programs, which can lead to practical and logistical issues.5,7 Health care systems and hospitals must proactively develop policies regarding the use of biosimilars.3 Further to the considerations of efficacy, safety, and cost-effectiveness that are standard in formulary reviews, the evaluation of biosimilars should include consideration of clinical, product, and institutional factors.3
An understanding of the theoretical decision framework used by prescribers when incorporating an innovation into practice can be helpful in understanding and improving the uptake of biosimilars.15 Previous research has indicated that 5 key criteria influence a clinician’s choice to implement an innovation: relative advantage (the degree to which the innovation is perceived as being better than its predecessor), compatibility (how consistent the innovation is with the existing values, past experiences, and needs of potential adopters), complexity (perception of difficulty of use), trialability (ability to experiment with the innovation on a limited basis before adoption), and observability (how observable the results are to others).15 Meeting these adoption criteria could positively influence the future success of biosimilars within an organization, and these criteria can also be used to evaluate an existing situation to determine steps needed for improvement.
The DTC of the authors’ organization is a multidisciplinary committee that makes decisions about medication listings in the drug formulary, as well as policies and procedures related to formulary processes. Drug utilization evaluation and stewardship pharmacists, who are part of the DTC, are responsible for conducting the formulary reviews.
To prepare for the addition of biosimilar drugs to the acute care formulary, steps were taken to educate pharmacy staff. This staff education took the form of a “backgrounder” document, a concise reference document designed to facilitate discussions between front-line pharmacy staff and the prescribers with whom they work on drug-use issues within the organization. The biosimilars backgrounder provided information about the development of biosimilars in Canada and related concepts, and discussed the non-interchangeability of biosimilars with reference biologics.
In 2016, the DTC decided that biosimilars listed on the provincial benefit list (for ambulatory drugs) would be provided to patients receiving care within the organization, for the purpose of continuity of care. This decision took into consideration the regulatory prohibition against interchanging a biosimilar with its originator product and was intended to prevent any disruptions in therapy for patients already stabilized on a branded product before admission to hospital. Further decisions about each biosimilar’s formulary status were then to be made through the usual review process.
To date, 3 biosimilar drugs have been added to the formulary, each with different approaches and considerations, as detailed in Table 2. All had undergone the Common Drug Review process, a national approach to reviewing drugs newly approved in Canada in order to make a recommendation on eligibility for public reimbursement,16 and were listed on the provincial drug benefit list before being considered for addition to the acute care formulary.
Table 2 Characteristics of 3 Biosimilars Introduced to a Health Authority’s Formulary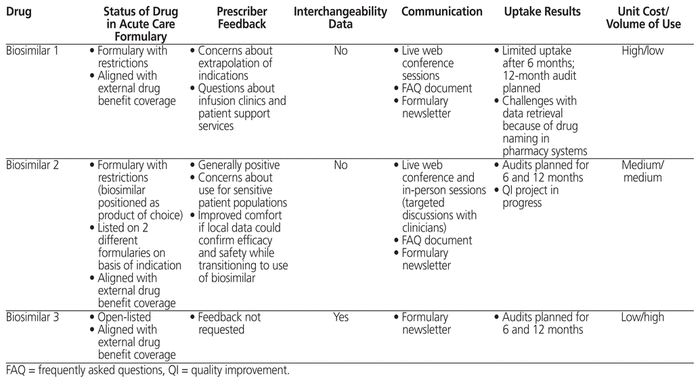
The first biosimilar was initially brought to market without approval for the full breadth of indications given for the originator brand. This situation arose from regulatory concerns about extrapolating data from one disease state to another because of differences in disease mechanisms and the safety profile of the biologic in different diseases. The indications not originally approved were added later, on the basis of additional data and rationales addressing concerns about mechanisms of action. Upon review of this biosimilar for formulary inclusion in the authors’ organization, the recommendation to the DTC and the subsequent decision by the DTC were that it be added with restrictions aligning it with the originator biologic on the formulary and with the criteria of the provincial drug benefit list for coverage. Stakeholder feedback was gathered from relevant clinicians within the organization. Through this process it was discovered that some prescribers were hesitant to use the biosimilar product because of questions about the regulatory process and a perceived lack of transparency concerning the scientific rationale used for approval of all indications through extrapolation. There was also uncertainty about patients’ access to infusion clinics for administration of the biologic. Although the brand had a well-established system of supports for administration of doses to patients, it was unclear how many administration sites the manufacturer of the biosimilar had confirmed.
Information about the formulary decision was disseminated via an electronic newsletter circulated to clinical staff, supplemented with live presentations (through online conference). A document of frequently asked questions (FAQs) was developed and linked to the formulary record. Uptake of the biosimilar was measured 6 months after its addition to the formulary. Results showed limited use, with unequal utilization patterns in various geographic areas of the province. Although the organization had listed the biosimilar on equal footing with the originator product, the added support services offered by the manufacturer of the originator brand made it difficult for the biosimilar to gain uptake. The auditing process also uncovered limitations with data capture in some pharmacy systems due to problems with drug nomenclature.
The situation for the second biosimilar was more complex because different indications had separate coverage sources, with the drug being listed on both the provincial acute care formulary and the formulary for a high-cost drug program. Additions to the formulary for the high-cost drug program follow a separate process for review and final decision.
Relevant prescribers were consulted about the proposed status of the biosimilar on the acute care formulary and the formulary for the high-cost drug program. Clinicians were generally receptive to using the biosimilar product for one indication; however, they expressed concerns about sensitive patient populations (i.e., patients in critical condition), in which no direct clinical studies had been done. There were also questions about potentially increased adverse effects of the biosimilar relative to the originator product, as reported in published studies. Prescribers noted that their confidence in using the biosimilar for sensitive patient populations would increase if local data (from within the organization) could be collected to assess efficacy and safety. In response to this request, a future quality improvement project was planned, with support from the drug utilization evaluation and stewardship pharmacist.
With a desire for stronger uptake of biosimilars, the DTC aimed to maximize use by positioning this biosimilar as the product of choice for the specified indications. This preference was communicated directly to stakeholders, who were consulted before broader communications to other clinical staff. An FAQ document was also developed for reference purposes. To determine uptake, utilization audits are planned for 6 and 12 months after implementation.
Supporting information for the third biosimilar included data from studies in which patients were switched from the originator product to the biosimilar, which provided a level of evidence not seen with the other 2 biosimilars. This third biosimilar was recommended by the Common Drug Review for reimbursement with conditions, and was added to the provincial drug benefit list as a regular benefit (“open listed”). Although the DTC wanted to increase uptake of the biosimilar, it took a cautious approach with this formulary listing, because there had been stakeholder disagreement with a previous DTC decision regarding the same class of drugs. With this biosimilar, the DTC recommended that it be listed with unrestricted formulary status, meaning that either the brand or the biosimilar product could be used in new starts, as well as for continuing therapy. Because the listing was at parity with the originator brand already on formulary, a broad stakeholder survey was not conducted; the same is often done for formulary decisions that represent line extensions (e.g., addition of new strengths of a drug that is already listed on the formulary) or changes to product listings that do not affect clinical practice. The decision was communicated to clinical staff through the electronic newsletter and live presentations. Utilization audits are planned for 6 and 12 months after implementation to measure uptake of the biosimilar.
In relation to the checklist proposed by Griffith and others,11 the organization’s process adequately addressed most considerations for formulary review of biosimilars. The areas of greatest challenge and focus were safety and efficacy, hospital and patient considerations, and economic considerations. In general, clinicians had some reservations about the available clinical data and questions about the sufficiency of data for the indications being considered for formulary inclusion. These reservations and questions were not surprising, given that the regulatory pathway and authorization of biosimilars for indications without supporting clinical studies represent a newer concept for prescribers.
Product naming was also identified as an important issue to be addressed. Having the ability to distinguish biosimilars from the corresponding originator products at order entry, for utilization reports, and for tracking of safety events (pharmacovigilance) is essential. In February 2019 (after this article had been accepted for publication), Health Canada issued formal guidance on how current and future biosimilars should be named,17 based on results from a joint public consultation conducted by Health Canada and the Institute for Safe Medication Practices Canada on this topic in 2018.18 In brief, Health Canada recommends that “biologic drugs, including biosimilars, will be identified by their unique brand name and non-proprietary (common) name, without the addition of a product-specific suffix.”17
Although the organization faced some challenges with the addition of biosimilars to the formulary, there were also positive aspects. For example, a process was put into place regarding the provision of biosimilars not yet added to formulary; such processes have been noted to support the entry of biosimilars into practice.3 The process included stakeholder feedback through targeted engagement of specific clinical groups, helping to ensure that key contacts were included in the discussion. Having active and direct involvement of the appropriate clinicians has been noted as an important component of formulary review of biosimilars.7 Also, efforts to support the education of pharmacy and clinical staff about DTC decisions were beneficial for disseminating knowledge about the rationale for biosimilar decisions.
A major consideration was non-interchangeability between biosimilars and their corresponding biologics, and the recognition that automatic interchange or substitution to another formulary item is not supported for biosimilars. However, as studies continue to evolve, with the inclusion of more trials with designs involving multiple switches between a biosimilar and its reference product, the immunogenicity effects of such practices will come to be better understood.4 Additionally, monitoring patient outcomes for efficacy and adverse events will be essential to confirm the effects of longer-term use of these agents.13 As learned during the formulary addition process, clinicians’ level of comfort with biosimilar products may be improved by collecting local data to confirm scientific data gathered in clinical trials of the biosimilar.
With respect to sustainability of the health care system, biosimilars have significant potential to reduce the costs associated with the biologics class of drugs.2,3,6,7 The cost savings associated with using biosimilars were a factor in their listing on formulary, but the overall impact for the organization is yet to be determined. The biosimilar examples described in this article had list prices ranging from 17% to 45% lower than the corresponding originator products. In the current economic climate, there is motivation to optimize drug expenditures and improve drug stewardship.
As drug experts, hospital pharmacists are ideally positioned to support the use of biosimilars.6 Pharmacist-led education initiatives can improve prescribers’ awareness of and comfort with biosimilars3 by decreasing knowledge gaps and misconceptions.19 A strong knowledge base of biosimilar concepts is necessary, and educational strategies to ensure that pharmacists have the necessary competency to select and recommend biosimilars should be in place early on. This requirement implies integration of biosimilars into pharmacy curriculums,19 as well as into continuing education for pharmacists in practice.
Our observations indicate that the uptake of biosimilars often begins in the acute care setting, as these agents may be started in hospital to stabilize a patient’s condition. As such, one vital consideration is alignment of the acute care formulary with public drug plans to ensure continuity of therapy for patients when they are moved from one care setting to another. Coordinating formulary listings for biosimilars in acute care and in the ambulatory setting will help to ensure that patients have timely access to these agents.
Our organization’s experience can be examined using the theoretical framework of 5 criteria that influence a prescriber’s decision to adopt innovations.15 Initially, the relative advantage of biosimilars was unclear. Compatibility was low, because of a lack of previous experience with biosimilars, whereas complexity was high, because the concepts surrounding biosimilars were new. The trialability and observability of biosimilars were also low, because the products could be used only after they had been added to the formulary. Uncertainty about biosimilars and a pre-existing affinity for the reference products yielded low motivation for clinicians to change their prescribing behaviour.15 However, involving clinicians in the review process and addressing knowledge gaps in the areas of clinical safety and efficacy were positive steps providing extra support for change.
The most salient issue was a lack of comfort and confidence on the part of prescribers, mainly related to the regulatory approval process allowing extrapolation of data from one disease condition to another. Some experts had the view that extrapolation should be guided only by appropriate clinical trial evidence. This uncertainty seemed to affect the acceptance of biosimilar 1 more than biosimilars 2 and 3. However, it was unclear whether other factors, such as it being the first biosimilar evaluated and the chronic nature of the disease state being treated, also contributed to slow and limited uptake. Biosimilar 2, which is typically used for shorter treatment periods, seems to have been better received. The approach with that biosimilar was structured to facilitate collection of local data, which enabled trialability and observability of the product’s effects in sensitive patient populations. Similar projects could be considered in the future, if capacity exists, as this support helps to address the compatibility of prescriber behaviour with the use of biosimilars.
Finally, it was observed that the approach to formulary review and inclusion may differ among various biosimilars, according to certain product-specific factors. A tailored approach that takes into account provincially based factors related to the sustainability, quality, and supporting infrastructure available for biologics is necessary for their success.20 To improve the availability of biosimilar drugs in the future, pharmacists and the DTC must apply appropriate formulary tools for practice management.7
1 Biosimilar drugs. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2017 Feb [cited 2018 Feb 10]. Available from: https://cadth.ca/sites/default/files/pdf/biosimilar_drugs_professional_en.pdf
2 Fact sheet: biosimilars. Ottawa (ON): Health Canada; 2017 Aug 3 [cited 2018 Feb 10]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/fact-sheet-biosimilars.html
3 Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P & T committees. P T. 2015;40(10):680–9.

4 Rifkin RM, Peck SR. Biosimilars: implications for clinical practice. J Oncol Pract. 2017;13(9 Suppl):24s–31s.
5 Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. Generics Biosimilars Initiat J. 2014;3(3):150–4.
6 Jarrett S, Dingermann T. Biosimilars are here: a hospital pharmacist’s guide to educating health care professionals on biosimilars. Hosp Pharm. 2015;50 (10):884–93.
7 Ventola CL. Biosimilars part 2: potential concerns and challenges for P&T committees. P T. 2013;38(6):329–35.
8 Hospital-based pharmacy and therapeutics committees: evolving responsibilities and membership. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2011 Jul [cited 2018 Feb 1]. Available from: https://www.cadth.ca/media/pdf/pharm-thera%20_com_es-23_e.pdf
9 Guidance document: information and submission requirements for biosimilar and biologic drugs. Ottawa (ON): Health Canada; 2016 Nov 14 [cited 2018 Feb 1]. Available from: https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2016-eng.pdf
10 Russell AS, Ahluwalla V, Barnabe C, Jamal S, Offer RC, Olsznski WP, et al. Subsequent entry biologics/biosimilars: a viewpoint from Canada. Clin Rheumatol. 2012;31(9):1289–92.
11 Griffith N, McBride A, Stevenson JG, Green L. Formulary selection criteria for biosimilars: considerations for US health-system pharmacists. Hosp Pharm. 2014;49(9):813–25.
12 Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70(22):2004–17.
13 Mckinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27–52.
14 FIP statement of policy. Pharmacist’s authority in pharmaceutical product selection: therapeutic interchange and substitution. The Hague (Netherlands): International Pharmaceutical Federation; 2018 Sep 2 [cited 2018 Oct 10]. Available from: https://fip.org/www/uploads/database_file.php?id=403&table_id=
15 Dylst P, Vulto A, Simoens S. Barriers to the uptake of biosimilars and possible solutions: a Belgian case study. Pharmacoeconomics. 2014;32(7):681–91.
16 CADTH common drug review (CDR). Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; [cited 2018 Feb 10]. Available from: https://www.cadth.ca/about-cadth/what-we-do/products-services/cdr
17 Notice to stakeholders - policy statement on the naming of biologic drugs. Ottawa (ON): Health Canada, Health Products and Food Branch, Biologics and Genetic Therapies Directorate, Office of Policy and International Collaboration; [modified 2019 Feb 14; cited 2019 March 21]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/biosimilar-biologic-notice-to-stakeholders-drugs-naming-of-biologics.html
18 Stakeholder consultation on the naming of biologic drugs. Toronto (ON): Institute of Safe Medication Practices Canada; [cited 2018 Feb 10]. Available from: https://www.ismp-canada.org/biosimilars/
19 Moorkens E, Vulto AG, Huys I, Dylst P, Godman B, Keuerleber S, et al. Policies for biosimilar uptake in Europe: an overview. PLoS One. 2017; 12(12):e0190147.
20 Towards an Alberta approach for biosimilar reimbursement. Summary report of the IHE Biosimilars Forum October 6, 2016. Edmonton (AB): Institute of Health Economics; 2016 [cited 2018 Feb 1]. Available from: https://www.ihe.ca/download/towards_an_alberta_approach_for_biosimilar_reimbursement.pdf
Competing interests: None declared. ( Return to Text )
Funding: None received. ( Return to Text )
Canadian Journal of Hospital Pharmacy, VOLUME 72, NUMBER 2, March-April 2019